Introduction
Hereditary hemorrhagic telangiectasia (HHT), formerly Osler-Weber-Rendu, is an inherited (autosomal dominant) disease that results in malformed blood vessels (see Image. Telangiectasia on the Tongue). The disease is named after the physicians who first independently described the condition: Henri Jules Louis Marie Rendu in 1896, William Osler in 1901, and Frederick Parkes Weber in 1907.[1] The malformations typically manifest as mucocutaneous telangiectasias and visceral arteriovenous malformations (AVMs).[2] These vascular malformations are responsible for much of the clinical bleeding associated with this disease, ranging from mild epistaxis to life-threatening intracranial bleeds.[3][4][5] Some patients with HHT develop pulmonary hypertension, a prothrombotic state, or immune dysfunction.[6] The earliest clinical sign of HHT, often occurring by the second decade of life, is recurrent epistaxis. Telangiectasias, which are dilated blood vessels, are frequently present on the skin and buccal mucosa in the third decade of life. The number of telangiectasias increases with age, accompanied by increased frequency of epistaxis or gastrointestinal (GI) bleeds, leading to anemia, poorer quality of life, and increased healthcare resource utilization, including iron or blood transfusions and hospitalizations.[7]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Genes comprise DNA, which encodes for distinct proteins that perform a specific function in the body. Through transcription, the DNA is converted into messenger RNA (mRNA) and then translated into a protein. HHT is an autosomal dominant disorder affecting blood vessels within multiple organ systems. The condition is suspected of haploinsufficiency, where 1 functional gene copy cannot produce the required protein to preserve function.[2] In 97% of patients with a definite clinical HHT diagnosis, a causative mutation is identified in 1 of 3 genes described below.[8] These mutations disrupt signaling by transforming growth factor-beta (TGF-B), essential for maintaining vascular integrity.[9][10][11]
HHT Type I (HHT1)
HHT1 stems from mutations in the gene ENG, which codes for endoglin on chromosome 9.[12] Endoglin is a membrane glycoprotein part of the tumor growth factor-beta (TGF-B) receptor complex needed for vascular integrity.
HHT Type 2 (HHT2)
HHT2 results from Activin A receptor-like type I (ACVRL1) mutations, which codes for the protein activin receptor-like kinase 1 (ALK1) on chromosome 12.[13] ALK1 is a type 1 cell-surface receptor for the TGF-B superfamily and is found on the surface of cells, particularly on the lining of developing arteries.[14]
HHT Associated with Juvenile Polyposis (JPHT or JP-HHT)
JPHT is due to a mutation in the gene Mothers Against Decapentaplegic homolog 4 (MADH4) that codes for the transcription factor SMAD4, a critical downstream effector of TGF-B signaling.[15] The mutations in JPHT are located on the last 4 exons of Smad4 and include several mutation types, including nonsense, missense, frameshift, and de novo.[6] The distribution in gene mutations among HHT patients is predominantly ENG (61%), followed by ACVRL1 (37%), and then MADH4 (2%). Over 600 mutations, including deletions, missense, nonsense, and insertions, have also been identified in ENG or ACVR genes.
Epidemiology
HHT affects 1 in 5,000-8,000 individuals and can affect both genders and people of all races.[2][5][6][7] Some studies have noted a higher incidence in women, although this gender difference may be attributed to access to healthcare resources.[16][17]
Pathophysiology
Pathogenesis of HHT is primarily due to the gene mutations (endoglin, ACVRL1, and SMAD4) that affect the endothelial cell receptors of the TGF-B superfamily. A mutation in these receptors prevents downstream signaling and disrupts angiogenesis, promoting a disorganized cytoskeleton and dysfunctional remodeling of the vascular endothelium. As a result, these vessels lose elasticity and remain chronically dilated.[18] The loss of vascular integrity, combined with constant pressure, results in telangiectasias (dilated microvessels) and large AVMs.[19][9] Due to the vessel walls' decreased elasticity and vascular lumen dilation, telangiectasias are fragile and more prone to hemorrhage. AVMs can form in the brain, lungs, GI tract, spine, or liver.[19] Up to 10% of HHT patients have cerebral AVMs, 15 to 45% develop pulmonary AVMs, and 75% have hepatic AVMs. Rupture of these AVMs can result in severe complications, including internal hemorrhage, embolic or hemorrhagic stroke, seizures, migraines, or brain abscesses.[17] Untreated pulmonary and hepatic AVMs can lead to arteriovenous shunting and pulmonary hypertension.[6]. There are phenotypic variations between HHT1 and HHT2. HHT1 patients develop epistaxis earlier in life and have pulmonary AVMs, while HHT2 patients are likelier to develop hepatic AVMs.[20]
Histopathology
Normally, arteries and veins are connected by an intermediary capillary system. AVMs and telangiectasias lack this intermediary, with a direct connection between the artery and the vein. Telangiectasias occur on mucocutaneous surfaces, while AVMs are found within internal organs. Histological evaluation of AVMs shows irregular endothelium, increased collagen and actin deposition in the basement membrane, and a disorganized basement membrane.[21]
History and Physical
Although autosomal dominant, the clinical manifestations of HHT are variable, even within families.[6] The classic triad consists of epistaxis, telangiectasias, and positive family history of similarly affected individuals.
Epistaxis
The earliest clinical manifestation and primary complaint in up to 96% of patients are spontaneous, recurrent epistaxis stemming from nasal mucosal telangiectasias. Epistaxis can occur as early as childhood and increases in prevalence with age, potentially resulting in anemia and the need for blood or iron transfusions. The average age of epistaxis onset is 12 years, with almost 100% of HHT patients affected by 40 years and an average of 18 monthly bleeds.[22][23] Evaluation of epistaxis should involve anterior rhinoscopy of the nasal cavity. If resources are available, nasal endoscopy or upper airway endoscopy with a rigid or flexible fiberoptic scope can be done to evaluate for the presence of telangiectasias in the nasal or oral mucosa (see Image. Telangiectasia in Nasal Cavity). However, great care needs to be taken to avoid unnecessary bleeding.
Mucocutaneous Telangiectasias
Telangiectasias of the skin and oral mucosa appear around the 3rd decade and increase in number and frequency with age.[6] Recurrent GI bleeds manifest later in 15-20% of HHT patients.[7] Patients should be queried about bleeding frequency and severity. On physical exam, HHT patients have telangiectasias involving the nasal mucosa, oral cavity including hard palate, tongue, and lips, as well as cutaneous lesions on the fingers and nose (see Images. HHT Telangiectasias on Thumb, Lip Telangiectasia).[9]
Cutaneous Telangiectasias on the Fingers
Visceral Arteriovenous Malformations (AVM)
Patients may also develop AVMs of the lungs, GI tract, brain, liver, or spine. AVM-related symptoms and complications should be assessed, including a history of stroke, heart failure, venous thromboembolism, iron deficiency, brain abscesses, arteriovenous shunting, liver disease, migraines, and pulmonary hypertension.[19] Cerebral and pulmonary AVMs typically form perinatally and pre-puberty, respectively.[24] In addition, because patients with the JPHT form are at higher odds of developing colon cancer, patients should be queried for signs or symptoms of colorectal cancer, such as weight loss, change in bowel habits, and history of colorectal cancer among affected family members. On exam, patients may present with hepatomegaly or hepatic bruits due to the left-to-right shunting of blood in hepatic AVMs.[25] Additionally, patients may exhibit pallor due to chronic anemia.
Family History
In patients with a positive family history of HHT, the presence of a visceral AVM essentially confirms the diagnosis since AVMs are rare in the general population. For unaffected patients with a parent with HHT, the disease cannot be ruled out due to variable age-related onset of signs and symptoms. European studies on HHT patients estimate the probability of clinical HHT in patients with an affected family member to range from 0.5, 0.22, and 0.01 at 0-, 16-, and 60 years.[7][26]
Classification Criteria
The diagnosis of HHT has relied on Curaçao Criteria, which encompasses the classic features of the disease: 1) spontaneous, recurrent epistaxis, 2) positive family history, 3) cutaneous or mucosal telangiectasias, and 4) visceral lesions. A definitive diagnosis is made if patients have 3 of the 4 criteria, and a possible or suspected diagnosis is made if patients meet 2 of the 4 criteria.[5] Of note is that the criteria have a poor negative predictive value in children under the age of 16 years.[27]
Evaluation
Genetic Testing
Genetic mutation testing should be done to confirm a diagnosis of HHT, including patients who meet 1-2 of the Curaçao criteria or young children with affected parents who are yet to develop the clinical manifestations.[19] Initial genetic testing should screen for the 3 most prevalent mutations: ENG, ACVRL1, and SMAD4. Testing should also be extended to family members. It is important to note that genetic mutations are not identified in up to 10 to 15% of HHT families, and a negative genetic test does not exclude the diagnosis of HHT. Once the diagnosis is confirmed, additional tests can be done to evaluate for other HHT manifestations. Screening should be done, regardless of a patient’s clinical symptoms, due to the danger of undiagnosed silent AVMs.
Ancillary Tests for Visceral AVMs
Pulmonary AVMs are often silent but can lead to strokes, massive hemoptysis, spontaneous hemothorax, transient ischemic attacks, and brain abscesses.[22] Sensitive screening tests to detect pulmonary AVMs include thoracic CT scan and transthoracic contrast/bubble echocardiography; both modalities can also detect pulmonary hypertension. Most screening protocols use contrast echocardiography as a first-line test, followed by a thoracic CT to determine the anatomic location and if embolization is viable. Chest X-rays, right-to-left shunt measurements, and blood oxygen measurements are less sensitive to identifying the presence of pulmonary AVMs.
Multiple cerebral AVMs are predictive of HHT.[28] The role of screening remains controversial due to the overall low risk, albeit significant morbidity or mortality, of hemorrhage. Additionally, the risks associated with treating asymptomatic cerebral AVMs potentially outweigh the benefits. An MRI brain with and without contrast is initially recommended for patients with cerebral symptoms, who have a known unstable cerebral aneurysm, or a family member who has had a cerebral hemorrhage since familial aneurysms have a higher risk of hemorrhage.[29] The gold standard for diagnosing and treating cerebral AVMs is diagnostic angiography, which carries a 0.5% risk of stroke.[25]
Routine gastrointestinal (GI) endoscopy is not typically performed. However, patients with anemia disproportionate to the severity of epistaxis or with a history of GI bleeding should undergo an esophagogastroduodenoscopy (EGD) to detect and treat GI AVMs.[9][22] If EGD is inconclusive, capsule endoscopy can be considered. Additionally, patients with the JPHT form of HHT should have a screening colonoscopy starting at the age of 15 due to a higher risk of colon cancer. This should be repeated every 3 years if no colon polyps are found. If polyps are detected, the patient should undergo yearly EGD and colonoscopy.[22] Screening for asymptomatic hepatic AVMs with Doppler ultrasonography is recommended because it is non-invasive and can improve patient management and outcomes. While Doppler ultrasonography is ideal due to its accuracy, cost, safety, and tolerability, depending on resources available and operator expertise, patients can be screened by alternate means, such as multiphase contrast CT or MRI.[19][6]
Laboratory Evaluation
Laboratory testing should be done before all surgical interventions, including a complete blood count and a type and screen or type and crossmatch.[9] Furthermore, all adults, regardless of symptoms and children with recurrent bleeding, should have annual complete blood count and ferritin levels measured to screen for iron deficiency anemia. If the patient is anemic but ferritin is normal, further workup with serum iron, transferrin saturation, and total iron-binding capacity should be performed.[19][22] HHT patients with severe epistaxis demonstrate microcytic iron deficiency anemia with low ferritin and elevated transferrin.[9]
Pregnancy
Pregnant women with HHT should have access to a multidisciplinary maternal-fetal team with knowledge of HHT, and pre-conception and prenatal diagnostic options should be discussed. Asymptomatic pregnant women should have an agitated saline transthoracic contrast echocardiography (TTCE) or a diagnostic low-dose chest CT without contrast for pulmonary AVMs. Any intervention should be delayed until the 2nd trimester.[22] Spinal AVMs can be detected with a spinal MRI and can be considered in pregnant women, particularly if epidural anesthesia is considered.[6] However, an expert consensus panel recommended against withholding an epidural, as risks of complications are unsubstantiated. Pregnant women with symptomatic cerebral AVMs or previous cerebral hemorrhage should have an unenhanced MRI in the second trimester. For asymptomatic cerebral AVMs, vaginal delivery may be attempted. However, for HHT patients with symptomatic cerebral AVMs or prior hemorrhage, a cesarean section should be considered to avoid the strain associated with delivering vaginally.[22]
Treatment / Management
Treatment options are tailored to the patient, and the best approach is based on local versus systemic measures. Due to the few randomized trials, there are no standard therapies for HHT. Management of HHT focuses on supportive care, preventing complications, and reducing symptom severity.
Epistaxis
Epistaxis prevention is the primary goal for HHT-related nosebleeds. Preventative measures include topical moisturizers and emollients, nasal hygiene with humidifiers and saline irrigations, and avoiding triggers and blood thinners.[30][19] Conservative management of ongoing epistaxis includes topical decongestant spray, manual pressure, absorbable nasal packing, and chemical cauterization with silver nitrate.[9] Non-dissolvable nasal packing should be avoided due to the risk of increased mucosal trauma with insertion and removal of the packing. Epistaxis refractory to conservative measures may require surgical or endovascular interventions.[19] Other medical interventions targeting the molecular biology of the disease have been used – many of which have gained and lost favor. These medical treatments aim to reduce nosebleeds' frequency, volume, and severity and improve quality of life. These topical and oral agents include estrogen agents (tamoxifen, raloxifene, and estriol ointment), tranexamic acid, thalidomide, beta-blockers (timolol or propranolol), and vascular endothelial growth factor (VEGF) inhibitors (bevacizumab).[9][31] Oral tranexamic acid is a possible option for treating HHT-epistaxis refractory to moisturizing topical therapies.[32] Two randomized control trials evaluating oral tranexamic acid for HHT-related epistaxis demonstrated a 17.3% reduction in epistaxis duration and a 54% reduction in epistaxis intensity.[33][34](A1)
Surgical interventions focused on prevention and reducing severity include electrosurgical plasma coagulation, potassium titanyl phosphate (KTP) laser photocoagulation, and sclerotherapy with sodium tetradecyl sulfate.[35] Laser photocoagulation improves the quality of life outcomes and decreases the frequency and severity of epistaxis. Other surgical interventions for moderate to severe epistaxis include septodermoplasty and Young’s procedure. Septodermoplasty involves removing the sinonasal mucosal and replacement with a split-thickness skin graft.[36] After the surgery, epistaxis is reduced for at least 2 years, though the problem typically recurs with time as telangiectasias affect the graft. This procedure has lost favor among many. Young’s technically reversible procedure involves the closure of 1 or both nostrils using mucocutaneous flaps, resulting in complete obstruction of airflow through the nose and resolution of epistaxis.[37] After Young’s procedure, patients suffer hyposmia and hypogeusia but also have complete cessation of epistaxis. This procedure is reserved for severe, life-threatening epistaxis and is relatively uncommon.(A1)
Anemia and Anticoagulation
All patients with iron deficiency and anemia require iron replacement, either orally or intravenously, if the oral form is not absorbed or well-tolerated. Oral replacement starts with 35 to 65 mg of elemental iron daily, taken 2 hours prior or 1 hour after meals. IV iron infusions may be required regularly, starting at 1 gm in a single dose or divided infusions for refractory or severe anemia.[38] Blood transfusions should be considered in the following situations: hemodynamic shock, comorbidities requiring a higher hemoglobin baseline before surgery or pregnancy, or inadequate hemoglobin levels despite iron transfusions. Anticoagulation is permissible in HHT patients.[39] Vitamin K antagonists and unfractionated and low-molecular-weight heparins are preferred and better than direct-acting oral anticoagulants (DOACs). Currently, there is more literature regarding tolerance with heparin and warfarin in HHT patients. Warfarin is the oral anticoagulant of choice because of its tolerance and the existence of a reversal agent. Finally, a small retrospective study evaluating DOAC showed increased HHT-related epistaxis while on DOACs.[40] If dual therapy is required, the duration of treatment should be minimized, and patients should be closely observed.(A1)
GI Bleeding
Patients with mild to moderate HHT-related GI bleeding can be managed with iron replacement. For severe cases, hemoglobin can be managed with scheduled iron replacement and transfusions. For patients with moderate to severe GI bleeding refractory to iron replacement and transfusions, there is moderate evidence for intravenous bevacizumab or other systemic antiangiogenic therapy and inadequate evidence for oral antifibrinolytics. Endoscopic argon plasma coagulation is an option to address bleeding and non-bleeding lesions during upper GI endoscopy; however, the evidence for this is low.[22]
Pulmonary AVMs (PAVMs)
Detection and treatment of asymptomatic PAVM are recommended due to associated neurological risks, including brain abscesses and paradoxical embolic strokes. The mainstay treatment of PAVMs is transcatheter embolization with embolic material such as metallic coils and Amplatzer vascular plugs.[25][22][41] A CT chest is recommended 3 to 6 months after the procedure to ensure recanalization of the occluded feeding artery did not occur. Surgery is reserved only for life-threatening hemorrhage. Similarly, lung transplantation is limited to patients with diffuse bilateral disease refractory to other treatment modalities. There is an association between oral microorganisms and PAVM-associated brain abscesses.[42] The pulmonary capillary bed filters small thrombi and bacteria that enter the bloodstream. Since the capillary bed is bypassed in PAVMs, a direct right-to-left shunt effectively forms, allowing paradoxical emboli to pass and cause brain abscesses or strokes.[43] Thus, antibiotic prophylaxis is recommended for any procedure that carries a risk of bacteremia, particularly dental procedures. Finally, patients with PAVMs should be followed long-term to detect the growth of untreated PAVMs and reperfusion of treated PAVMS.
Hepatic AVMs (HAVMs)
Management of HAVMs is based on the symptoms and type of complications. HHT patients with HAVMs may have high-output cardiac failure, portal hypertension, or cirrhosis, which are managed medically. High-output cardiac failure is treated with blood transfusions for anemia, salt and fluid restriction, beta-blockers, and diuretics. Intravenous bevacizumab can be considered for patients with high-output cardiac failure who fail initial management. Portal hypertension and cirrhosis are treated with salt and fluid restriction, diuretics, and paracentesis. Liver transplantation is reserved for those with symptomatic HAVMs who are refractory to medical management.[44] Finally, hepatic artery embolization should be avoided since the procedure is only temporizing and carries high morbidity and mortality.(B2)
Cerebral AVMs (CAVMs)
HHT-related CAVMs are low-grade, usually small, and cortically located with superficial venous drainage. Patients symptomatic from CAVMs should be referred to a center with neurovascular expertise. The natural history of CAVMs associated with HHT is slightly more favorable than sporadic AVMs, with a yearly rupture rate of 1.3% vs. 2.2%.[45][46] As such, conservative management should be considered for CAVMs in HHT patients. Furthermore, although there are advances in interventional therapies for CAVMs, the risks still outweigh these benefits. The ARUBA trial evaluated interventional versus medical therapy for 223 non-HHT patients with CAVMs and found the risk of stroke or death to be 3 times higher (30.7% vs. 10.1%) in the interventional arm.[46] For HHT patients with symptomatic CAVMs or risk factors, such as a family history of cerebral hemorrhage, treatment options are embolization, microsurgery, stereotactic radiation, or a combination of these modalities. Microsurgery for low-grade lesions (Spetzler-Martin grade I or II) can be effective. Spetzler-Martin grading scale estimates the risk of open surgery for cerebral AVMs. A grade 1 AVM describes a small, superficial lesion located in a non-critical area of the brain and is considered low risk for surgery. A grade 6 AVM is non-operable.[47] One series demonstrated a 100% obliteration rate with a 3.2% rate of neurologic deficits and 0% mortality with microsurgery.[48] Gamma knife surgery also has a similar risk profile with 100% obliteration rates for lesions < 1 mL in volume. For CAVMs that are poor surgical candidates, stereotactic radiosurgery can be considered but has a lower cure rate for larger lesions.[49] Embolization alone is ineffective in addressing CAVMs but could be a helpful adjunct with surgery or stereotactic radiation.(A1)
Differential Diagnosis
Bleeding from diseases like von Willebrand disease or hemophilia is more generalized and occurs in an injury setting. In contrast, bleeding from HHT is more localized to the malformed blood vessels. Several diseases share similar clinical manifestations to HHT and must be ruled out during the workup. Referral to a hematologist can prove very helpful.
Limited Systemic Sclerosis
Patients with limited systemic sclerosis, called CREST syndrome, develop calcinosis, Raynaud disease, esophageal dysmotility, sclerodactyly, and telangiectasias. However, recurrent epistaxis is not a common feature of the syndrome. Specific autoantibodies may be positive in scleroderma, including anti-centromere antibodies, anti-topoisomerase I (Scl-70), and anti-RNA polymerase III.[50]
Ataxia-Telangiectasia
Ataxia-telangiectasia is an autosomal recessive condition that presents with cerebellar atrophy with progressive ataxia, cutaneous telangiectasias, immune defects, and increased risk for malignancies. Symptoms typically occur in the first or second decade of life. Key features of this condition include elevated serum alpha-fetoprotein (AFP) and decreased total IgG and IgA.[51]
Generalized Essential Telangiectasia
Generalized essential telangiectasia is rare; inherited and sporadic cases have been reported. The telangiectasias first appear on the lower extremities and slowly spread to involve the entire body.[52]
Hereditary Benign Telangiectasia
Hereditary benign telangiectasia is an autosomal dominant primary telangiectasia disorder with the development of telangiectasias on the skin and lips during birth or childhood. Lesions are usually asymptomatic and do not have systemic involvement. Unlike in HHT, histology of hereditary benign telangiectasia demonstrates preserved skin and dilated vessels with thicker capillary walls, explaining the lack of hemorrhage.[53]
Rosacea
Rosacea is a common skin disorder with recurrent facial flushing, erythema, telangiectasias, and inflammatory pustules on the face. Etiological factors for the development of the condition include genetics, environmental factors, neurovascular deregulation, and microorganisms.[54] See Image. Ocular Telangiectasia.
Telangiectasia Macularis Eruptiva Perstans
Telangiectasia macularis eruptiva perstans is a rare form of mastocytosis. The condition is seen more frequently in adults. Telangiectatic macules manifest as flat, reddish-brown lesions on the skin due to infiltration of mast cells into the upper dermis. Systemic involvement may involve the bone marrow, GI tract, liver, and lymph.[55]
Dermatomyositis
Dermatomyositis is an idiopathic chronic inflammatory autoimmune disorder of the skin and muscles. Skin manifestations include heliotrope rash around the eyes, papules over digits, and periungual telangiectasias.[56]
Systemic Lupus Erythematosus (SLE)
Lupus erythematosus is a multi-organ autoimmune disease with varying clinical presentations. Cutaneous manifestations include a malar rash, oral and nasal ulcers, and periungual telangiectasias.[57]
Radiation Oncology
As discussed above, stereotactic radiation therapy has been used to manage cerebral AVMs as a single modality or in conjunction with microsurgery or embolization for larger lesions.[58] Gamma knife radiosurgery offers obliteration rates of 100% for AVMs smaller than 1 mL, 85% for 1 to 4 mL, and 58% for lesions > 4 mL. The risk of symptomatic radiation necrosis directly correlates with increasing AVM size.[59]
Medical Oncology
Dysfunctional angiogenesis is thought to play a role in the pathogenesis of HHT, and research has focused on whether anti-angiogenic substances and immunomodulatory agents can effectively treat HHT. Bevacizumab is a humanized recombinant monoclonal antibody against vascular endothelial growth factor (VEGF), elevated in HHT. VEGF stimulates arterial, lymphatic, and venous development and suppresses endothelial cell apoptosis. Elevated VEGF levels can result in immature, abnormal vessel formation with constant remodeling.[60] Clinical trials and studies have explored the effectiveness of the VEGF inhibitor, administered systemically, topically, or submucosally, for control of HHT-related epistaxis.[61][62][63][64][65] Double-blind, randomized control trials and other comparative studies evaluating topical and intranasal submucosal bevacizumab demonstrate mixed conclusions when evaluating the severity of HHT-related epistaxis.[66] Two studies evaluating the submucosal application of bevacizumab demonstrated either a trend toward or significant improvement in epistaxis severity.[67]
Primary risks associated with topical or submucosal injection of bevacizumab include septal perforation and osteonecrosis. In 1 clinical trial, systemic therapy with bevacizumab (5 mg/kg IV every 2 weeks for 6 treatments) demonstrated reduced epistaxis episodes and high cardiac output.[61] Side effects of systemic bevacizumab at oncologic doses (5 to 15 mg/kg) include GI perforation, hemorrhage, thromboembolism, poor wound healing, and reversible posterior leukoencephalopathy, which were not noted in the lower doses used to treat HHT. However, common adverse effects included hypertension, nausea, diarrhea, headache, and muscle or abdominal pain when used systemically. Tacrolimus, a calcineurin inhibitor used for immunosuppression, has been shown to activate the ALK1-SMAD1/5/8 pathway and improve defects due to ALK1 loss. Thalidomide, an immunomodulatory imide drug, has been shown to downregulate VEGF levels in HHT, improve the integrity of vessel walls, and reduce epistaxis.[68][69][70] However, adverse effects, including neuropathy and fatigue, have limited its use as a therapeutic option.[32] Studies have evaluated selective estrogen response modifiers (raloxifene, tamoxifen), progestins, and estrogens to reduce epistaxis and GI bleeding; however, findings are inconsistent.[32][71] Furthermore, potential side effects of hormone therapy, including gynecomastia, weight gain, and venous thromboembolism, have prevented its wide use as a treatment option.
Prognosis
The literature evaluating the overall survival and prognosis of HHT is limited but may have a shorter expected lifespan. A retrospective study demonstrated a lower median age of death in HHT versus non-HHT patients (63.2 vs. 70.0 years).[72] A more recent prospective study (n=675) found HHT patients to have poorer survival compared to matched controls, with a median age of death of 77 years versus 80 years.[17] Furthermore, the study found hazard ratios for death were highest within the first 3 years after HHT diagnosis and subsequently decreased. This decrease in HR could explain how HHT may be diagnosed after an acute complication such as a stroke. Early detection and screening of HHT and preventing complications could help increase life expectancy.
Complications
HHT patients have a higher risk of bleeding and neurologic complications, including anemia, cerebral abscess, stroke, venous thrombosis, and heart failure.[17] [Table 1]
Table 1. Specific Complications Found in HHT and Recommended Prevention and Treatment
| Complication | Treatment/Prevention |
| Iron Deficiency | Annual screen with complete blood count, ferritin,
total iron-binding capacity [TIBC], and reticulocyte count Replete iron either via oral or IV when ferritin is less than 50 |
| Cerebral Abscess |
Screen and embolization of pulmonary AVMs Infection prophylaxis with antibiotics before procedures |
| Paradoxical Stroke |
Screen and embolization of pulmonary AVMs Replete iron either via oral or IV when ferritin is less than 50 Iron deficiency seems to increase the risk of thrombotic events |
| Venous Thrombosis |
Replete iron either via oral or IV when ferritin is less than 50 Iron deficiency seems to increase the risk of thrombotic events Anticoagulation is not contraindicated but has been discussed with specialists |
| Heart Failure |
Check for annual brain natriuretic peptide (BNP) in the presence of Hepatic AVMs |
Postoperative and Rehabilitation Care
Scarring is a common complication after laser ablation or electrosurgical cauterization of intranasal telangiectasias due to loss of mucociliary clearance and subsequent crusting. Postoperative nasal hygiene with topical saline sprays and irrigations and deliberate postoperative debridement of crusts can prevent scarring and synechiae formation. There are no definitive guidelines for optimal postoperative management for AVMs after endovascular intervention or surgery. Postoperative care, including activity restrictions, is based on the location of the AVM, size, the number of lesions, type of procedure (endovascular versus open), and extent of surgery.[73] Patients undergoing microsurgery for cerebral AVMs may require physical therapy after the procedure. Additional imaging may be necessary to assess the embolization site and recurrence.[74]
Consultations
Since HHT affects multiple organ systems, adequate treatment requires multidisciplinary care. A referral to a hematologist is indicated to manage iron deficiency anemia and for anticoagulation if diagnosed with venous thromboembolism. Consider referring to an otolaryngologist for the management of epistaxis. A gastroenterologist should be involved if there is a concern for GI bleeds. HHT patients with pulmonary AVMs should be seen by a pulmonologist, interventional radiologist, or thoracic surgeon. The presence of a cerebral AVM warrants referral to a neurosurgeon. If embolization is not an option, an interventional radiologist or general surgeon should manage hepatic AVMs.
Deterrence and Patient Education
Affected individuals must receive education and counseling on the implications of a possible or definitive diagnosis of HHT. Given that HHT is an autosomal dominant disease, it is essential to screen family members and provide genetic counseling before conception. Early diagnosis before patients are symptomatic can allow for screening tests and interventions to be done promptly and assist in diagnosing at-risk family members.[25]
Pearls and Other Issues
Early diagnosis of HHT is based on clinical findings. Genetic testing helps confirm the diagnosis but is not required for an index case unless 1) they are children who have not developed all the clinical features or 2) to test at-risk family members and identify a family-specific mutation. Screening and treatment of visceral AVMs depend on the potential for high-risk complications. For instance, proactive screening and treating asymptomatic pulmonary AVMs are encouraged compared to asymptomatic hepatic or cerebral AVMs.[25]
Enhancing Healthcare Team Outcomes
HHT can affect multiple organ systems, and early recognition and screening for clinical manifestations and prompt interventions can decrease morbidity and mortality. Epistaxis is one of the most common presenting symptoms. Patients with HHT benefit from a multidisciplinary approach and should ideally be referred to a center specializing in HHT.
Media
(Click Image to Enlarge)

Telangiectasia in Nasal Cavity. Rigid nasal endoscopy of the right nasal cavity demonstrating telangiectasias in hereditary hemorrhagic telangiectasia.
Contributed by T Locke, MD
(Click Image to Enlarge)
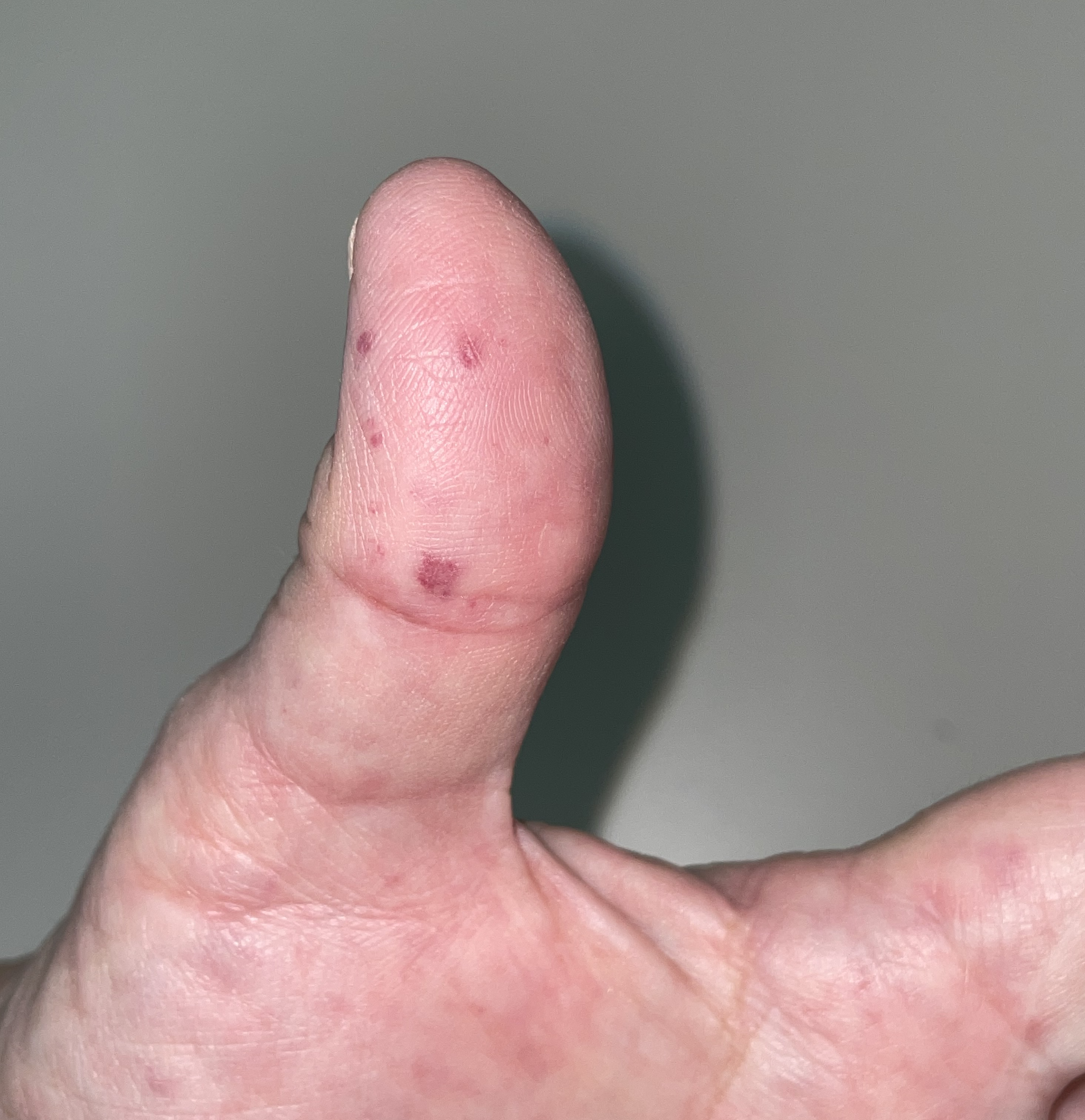
Hereditary Hemorrhagic Telangiectasias on Thumb
Contributed by P Chen, MD
(Click Image to Enlarge)
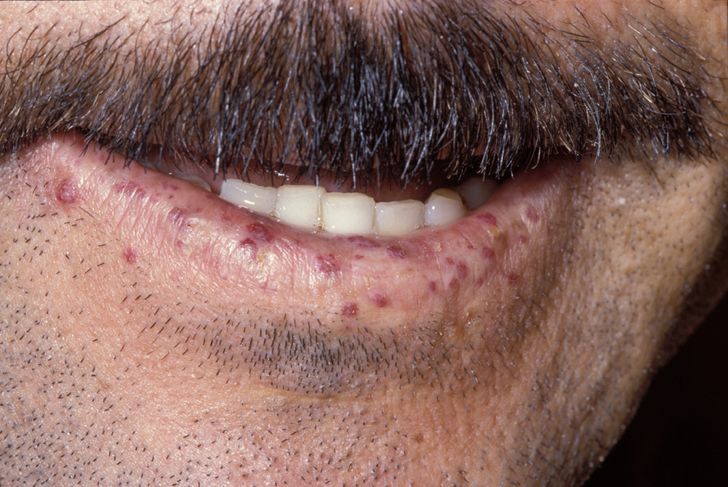
Lip Telangiectasia. The image depicts the presentation of a patient with hereditary hemorrhagic telangiectasia.
Herbert L Fred, MD, Hendrik A van Dijk, Public Domain, via Wikimedia Commons
(Click Image to Enlarge)
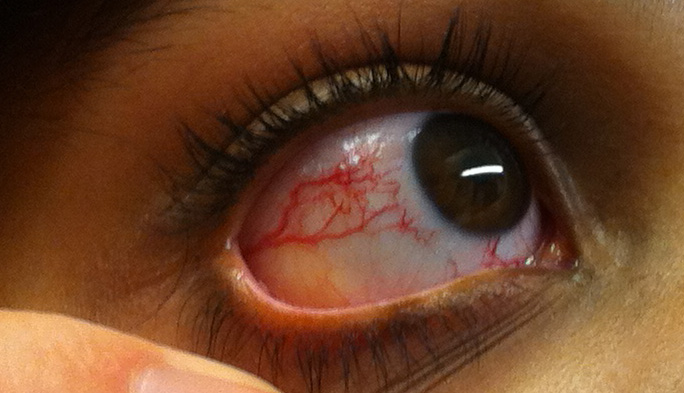
Ocular Telangiectasia. This image portrays the prominent ocular telangiectasia that can be seen in some people with this condition.
Thomas O Crawford, Public Domain, via Wikimedia Commons
(Click Image to Enlarge)

Telangiectasia on the Tongue. This patient has hereditary hemorrhagic telangiectasia, formerly known as Osler-Weber-Rendu syndrome, a multisystem vascular genetic disorder that produces blood vessel malformations.
Contributed by RE Sumpter, Public Domain, via Public Health Image Library
References
Kritharis A, Al-Samkari H, Kuter DJ. Hereditary hemorrhagic telangiectasia: diagnosis and management from the hematologist's perspective. Haematologica. 2018 Sep:103(9):1433-1443. doi: 10.3324/haematol.2018.193003. Epub 2018 May 24 [PubMed PMID: 29794143]
Level 3 (low-level) evidenceHalderman AA, Ryan MW, Clark C, Sindwani R, Reh DD, Poetker DM, Invernizzi R, Marple BF. Medical treatment of epistaxis in hereditary hemorrhagic telangiectasia: an evidence-based review. International forum of allergy & rhinology. 2018 Jun:8(6):713-728. doi: 10.1002/alr.22094. Epub 2018 Feb 2 [PubMed PMID: 29393992]
Jackson SB, Villano NP, Benhammou JN, Lewis M, Pisegna JR, Padua D. Gastrointestinal Manifestations of Hereditary Hemorrhagic Telangiectasia (HHT): A Systematic Review of the Literature. Digestive diseases and sciences. 2017 Oct:62(10):2623-2630. doi: 10.1007/s10620-017-4719-3. Epub 2017 Aug 23 [PubMed PMID: 28836046]
Level 1 (high-level) evidenceMorgan T, McDonald J, Anderson C, Ismail M, Miller F, Mao R, Madan A, Barnes P, Hudgins L, Manning M. Intracranial hemorrhage in infants and children with hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome). Pediatrics. 2002 Jan:109(1):E12 [PubMed PMID: 11773580]
Level 3 (low-level) evidenceShovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). American journal of medical genetics. 2000 Mar 6:91(1):66-7 [PubMed PMID: 10751092]
Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. European journal of human genetics : EJHG. 2009 Jul:17(7):860-71. doi: 10.1038/ejhg.2009.35. Epub 2009 Apr 1 [PubMed PMID: 19337313]
Plauchu H, de Chadarévian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. American journal of medical genetics. 1989 Mar:32(3):291-7 [PubMed PMID: 2729347]
Level 2 (mid-level) evidenceMcDonald J, Wooderchak-Donahue W, VanSant Webb C, Whitehead K, Stevenson DA, Bayrak-Toydemir P. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Frontiers in genetics. 2015:6():1. doi: 10.3389/fgene.2015.00001. Epub 2015 Jan 26 [PubMed PMID: 25674101]
Sautter NB, Smith TL. Treatment of Hereditary Hemorrhagic Telangiectasia-Related Epistaxis. Otolaryngologic clinics of North America. 2016 Jun:49(3):639-54. doi: 10.1016/j.otc.2016.02.010. Epub [PubMed PMID: 27267016]
Lesca G, Burnichon N, Raux G, Tosi M, Pinson S, Marion MJ, Babin E, Gilbert-Dussardier B, Rivière S, Goizet C, Faivre L, Plauchu H, Frébourg T, Calender A, Giraud S, French Rendu-Osler Network. Distribution of ENG and ACVRL1 (ALK1) mutations in French HHT patients. Human mutation. 2006 Jun:27(6):598 [PubMed PMID: 16705692]
Lesca G, Genin E, Blachier C, Olivieri C, Coulet F, Brunet G, Dupuis-Girod S, Buscarini E, Soubrier F, Calender A, Danesino C, Giraud S, Plauchu H, French-Italian HHT Network. Hereditary hemorrhagic telangiectasia: evidence for regional founder effects of ACVRL1 mutations in French and Italian patients. European journal of human genetics : EJHG. 2008 Jun:16(6):742-9. doi: 10.1038/ejhg.2008.3. Epub 2008 Feb 20 [PubMed PMID: 18285823]
McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nature genetics. 1994 Dec:8(4):345-51 [PubMed PMID: 7894484]
Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nature genetics. 1996 Jun:13(2):189-95 [PubMed PMID: 8640225]
Azuma H. Genetic and molecular pathogenesis of hereditary hemorrhagic telangiectasia. The journal of medical investigation : JMI. 2000 Aug:47(3-4):81-90 [PubMed PMID: 11019486]
Level 3 (low-level) evidenceGallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet (London, England). 2004 Mar 13:363(9412):852-9 [PubMed PMID: 15031030]
Donaldson JW, McKeever TM, Hall IP, Hubbard RB, Fogarty AW. The UK prevalence of hereditary haemorrhagic telangiectasia and its association with sex, socioeconomic status and region of residence: a population-based study. Thorax. 2014 Feb:69(2):161-7. doi: 10.1136/thoraxjnl-2013-203720. Epub 2013 Nov 4 [PubMed PMID: 24188926]
Donaldson JW, McKeever TM, Hall IP, Hubbard RB, Fogarty AW. Complications and mortality in hereditary hemorrhagic telangiectasia: A population-based study. Neurology. 2015 May 5:84(18):1886-93. doi: 10.1212/WNL.0000000000001538. Epub 2015 Apr 10 [PubMed PMID: 25862798]
Fernández-L A, Sanz-Rodriguez F, Blanco FJ, Bernabéu C, Botella LM. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clinical medicine & research. 2006 Mar:4(1):66-78 [PubMed PMID: 16595794]
Level 3 (low-level) evidenceGrigg C, Anderson D, Earnshaw J. Diagnosis and Treatment of Hereditary Hemorrhagic Telangiectasia. Ochsner journal. 2017 Summer:17(2):157-161 [PubMed PMID: 28638289]
Dupuis-Girod S, Bailly S, Plauchu H. Hereditary hemorrhagic telangiectasia: from molecular biology to patient care. Journal of thrombosis and haemostasis : JTH. 2010 Jul:8(7):1447-56. doi: 10.1111/j.1538-7836.2010.03860.x. Epub 2010 Mar 19 [PubMed PMID: 20345718]
Level 3 (low-level) evidenceDuncan BW, Kneebone JM, Chi EY, Hraska V, Isik FF, Rosenthal GL, Jones TK, Starnes SL, Lupinetti FM. A detailed histologic analysis of pulmonary arteriovenous malformations in children with cyanotic congenital heart disease. The Journal of thoracic and cardiovascular surgery. 1999 May:117(5):931-8 [PubMed PMID: 10220688]
Level 3 (low-level) evidenceFaughnan ME, Mager JJ, Hetts SW, Palda VA, Lang-Robertson K, Buscarini E, Deslandres E, Kasthuri RS, Lausman A, Poetker D, Ratjen F, Chesnutt MS, Clancy M, Whitehead KJ, Al-Samkari H, Chakinala M, Conrad M, Cortes D, Crocione C, Darling J, de Gussem E, Derksen C, Dupuis-Girod S, Foy P, Geisthoff U, Gossage JR, Hammill A, Heimdal K, Henderson K, Iyer VN, Kjeldsen AD, Komiyama M, Korenblatt K, McDonald J, McMahon J, McWilliams J, Meek ME, Mei-Zahav M, Olitsky S, Palmer S, Pantalone R, Piccirillo JF, Plahn B, Porteous MEM, Post MC, Radovanovic I, Rochon PJ, Rodriguez-Lopez J, Sabba C, Serra M, Shovlin C, Sprecher D, White AJ, Winship I, Zarrabeitia R. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Annals of internal medicine. 2020 Dec 15:173(12):989-1001. doi: 10.7326/M20-1443. Epub 2020 Sep 8 [PubMed PMID: 32894695]
AAssar OS, Friedman CM, White RI Jr. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. The Laryngoscope. 1991 Sep:101(9):977-80 [PubMed PMID: 1886446]
Shovlin CL, Jackson JE, Bamford KB, Jenkins IH, Benjamin AR, Ramadan H, Kulinskaya E. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax. 2008 Mar:63(3):259-66 [PubMed PMID: 17981912]
Garg N, Khunger M, Gupta A, Kumar N. Optimal management of hereditary hemorrhagic telangiectasia. Journal of blood medicine. 2014:5():191-206. doi: 10.2147/JBM.S45295. Epub 2014 Oct 15 [PubMed PMID: 25342923]
Shovlin CL, Hughes JM, Tuddenham EG, Temperley I, Perembelon YF, Scott J, Seidman CE, Seidman JG. A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nature genetics. 1994 Feb:6(2):205-9 [PubMed PMID: 8162076]
Pahl KS, Choudhury A, Wusik K, Hammill A, White A, Henderson K, Pollak J, Kasthuri RS. Applicability of the Curaçao Criteria for the Diagnosis of Hereditary Hemorrhagic Telangiectasia in the Pediatric Population. The Journal of pediatrics. 2018 Jun:197():207-213. doi: 10.1016/j.jpeds.2018.01.079. Epub 2018 Apr 11 [PubMed PMID: 29655863]
Bharatha A, Faughnan ME, Kim H, Pourmohamad T, Krings T, Bayrak-Toydemir P, Pawlikowska L, McCulloch CE, Lawton MT, Dowd CF, Young WL, Terbrugge KG. Brain arteriovenous malformation multiplicity predicts the diagnosis of hereditary hemorrhagic telangiectasia: quantitative assessment. Stroke. 2012 Jan:43(1):72-8. doi: 10.1161/STROKEAHA.111.629865. Epub 2011 Oct 27 [PubMed PMID: 22034007]
Shovlin CL, Sodhi V, McCarthy A, Lasjaunias P, Jackson JE, Sheppard MN. Estimates of maternal risks of pregnancy for women with hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): suggested approach for obstetric services. BJOG : an international journal of obstetrics and gynaecology. 2008 Aug:115(9):1108-15. doi: 10.1111/j.1471-0528.2008.01786.x. Epub 2008 May 30 [PubMed PMID: 18518871]
Level 2 (mid-level) evidenceTunkel DE, Anne S, Payne SC, Ishman SL, Rosenfeld RM, Abramson PJ, Alikhaani JD, Benoit MM, Bercovitz RS, Brown MD, Chernobilsky B, Feldstein DA, Hackell JM, Holbrook EH, Holdsworth SM, Lin KW, Lind MM, Poetker DM, Riley CA, Schneider JS, Seidman MD, Vadlamudi V, Valdez TA, Nnacheta LC, Monjur TM. Clinical Practice Guideline: Nosebleed (Epistaxis). Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery. 2020 Jan:162(1_suppl):S1-S38. doi: 10.1177/0194599819890327. Epub [PubMed PMID: 31910111]
Level 1 (high-level) evidenceYaniv E, Preis M, Shevro J, Nageris B, Hadar T. Anti-estrogen therapy for hereditary hemorrhagic telangiectasia - a long-term clinical trial. Rhinology. 2011 Jun:49(2):214-6. doi: 10.4193/Rhino09.201. Epub [PubMed PMID: 21743879]
Level 1 (high-level) evidenceAl-Samkari H. Hereditary hemorrhagic telangiectasia: systemic therapies, guidelines, and an evolving standard of care. Blood. 2021 Feb 18:137(7):888-895. doi: 10.1182/blood.2020008739. Epub [PubMed PMID: 33171488]
Gaillard S, Dupuis-Girod S, Boutitie F, Rivière S, Morinière S, Hatron PY, Manfredi G, Kaminsky P, Capitaine AL, Roy P, Gueyffier F, Plauchu H, ATERO Study Group. Tranexamic acid for epistaxis in hereditary hemorrhagic telangiectasia patients: a European cross-over controlled trial in a rare disease. Journal of thrombosis and haemostasis : JTH. 2014 Sep:12(9):1494-502. doi: 10.1111/jth.12654. Epub 2014 Jul 29 [PubMed PMID: 25040799]
Level 1 (high-level) evidenceGeisthoff UW, Seyfert UT, Kübler M, Bieg B, Plinkert PK, König J. Treatment of epistaxis in hereditary hemorrhagic telangiectasia with tranexamic acid - a double-blind placebo-controlled cross-over phase IIIB study. Thrombosis research. 2014 Sep:134(3):565-71. doi: 10.1016/j.thromres.2014.06.012. Epub 2014 Jun 16 [PubMed PMID: 25005464]
Level 1 (high-level) evidenceBoyer H, Fernandes P, Le C, Yueh B. Prospective randomized trial of sclerotherapy vs standard treatment for epistaxis due to hereditary hemorrhagic telangiectasia. International forum of allergy & rhinology. 2015 May:5(5):435-40. doi: 10.1002/alr.21484. Epub 2015 Feb 2 [PubMed PMID: 25643928]
Level 1 (high-level) evidenceKuan EC, Peng KA, Thompson CF, Suh JD, Wang MB. Sinonasal quality of life outcomes following laser treatment of epistaxis related to hereditary hemorrhagic telangiectasia. Lasers in medical science. 2017 Apr:32(3):527-531. doi: 10.1007/s10103-017-2144-7. Epub 2017 Jan 24 [PubMed PMID: 28116537]
Level 2 (mid-level) evidenceLund VJ, Darby Y, Rimmer J, Amin M, Husain S. Nasal closure for severe hereditary haemorrhagic telangiectasia in 100 patients. The Lund modification of the Young's procedure: a 22-year experience. Rhinology. 2017 Jun 1:55(2):135-141. doi: 10.4193/Rhin16.315. Epub [PubMed PMID: 28064338]
Peyrin-Biroulet L, Williet N, Cacoub P. Guidelines on the diagnosis and treatment of iron deficiency across indications: a systematic review. The American journal of clinical nutrition. 2015 Dec:102(6):1585-94. doi: 10.3945/ajcn.114.103366. Epub 2015 Nov 11 [PubMed PMID: 26561626]
Level 1 (high-level) evidenceEdwards CP, Shehata N, Faughnan ME. Hereditary hemorrhagic telangiectasia patients can tolerate anticoagulation. Annals of hematology. 2012 Dec:91(12):1959-68. doi: 10.1007/s00277-012-1553-8. Epub 2012 Sep 30 [PubMed PMID: 23053175]
Level 2 (mid-level) evidenceShovlin CL, Millar CM, Droege F, Kjeldsen A, Manfredi G, Suppressa P, Ugolini S, Coote N, Fialla AD, Geisthoff U, Lenato GM, Mager HJ, Pagella F, Post MC, Sabbà C, Sure U, Torring PM, Dupuis-Girod S, Buscarini E, VASCERN-HHT. Safety of direct oral anticoagulants in patients with hereditary hemorrhagic telangiectasia. Orphanet journal of rare diseases. 2019 Aug 28:14(1):210. doi: 10.1186/s13023-019-1179-1. Epub 2019 Aug 28 [PubMed PMID: 31462308]
Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, Spears J, Brown DH, Buscarini E, Chesnutt MS, Cottin V, Ganguly A, Gossage JR, Guttmacher AE, Hyland RH, Kennedy SJ, Korzenik J, Mager JJ, Ozanne AP, Piccirillo JF, Picus D, Plauchu H, Porteous ME, Pyeritz RE, Ross DA, Sabba C, Swanson K, Terry P, Wallace MC, Westermann CJ, White RI, Young LH, Zarrabeitia R, HHT Foundation International - Guidelines Working Group. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. Journal of medical genetics. 2011 Feb:48(2):73-87. doi: 10.1136/jmg.2009.069013. Epub 2009 Jun 23 [PubMed PMID: 19553198]
Shovlin C, Bamford K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. British dental journal. 2008 Nov 22:205(10):531-3. doi: 10.1038/sj.bdj.2008.978. Epub [PubMed PMID: 19023305]
Nam TK, Park YS, Kwon JT. Brain Abscesses Associated with Asymptomatic Pulmonary Arteriovenous Fistulas. Journal of Korean Neurosurgical Society. 2017 Jan 1:60(1):118-124. doi: 10.3340/jkns.2015.0707.023. Epub 2016 Dec 29 [PubMed PMID: 28061502]
Lerut J, Orlando G, Adam R, Sabbà C, Pfitzmann R, Klempnauer J, Belghiti J, Pirenne J, Thevenot T, Hillert C, Brown CM, Gonze D, Karam V, Boillot O, European Liver Transplant Association. Liver transplantation for hereditary hemorrhagic telangiectasia: Report of the European liver transplant registry. Annals of surgery. 2006 Dec:244(6):854-62; discussion 862-4 [PubMed PMID: 17122610]
Level 2 (mid-level) evidenceYang W, Liu A, Hung AL, Braileanu M, Wang JY, Caplan JM, Colby GP, Coon AL, Tamargo RJ, Ahn ES, Huang J. Lower Risk of Intracranial Arteriovenous Malformation Hemorrhage in Patients With Hereditary Hemorrhagic Telangiectasia. Neurosurgery. 2016 May:78(5):684-93. doi: 10.1227/NEU.0000000000001103. Epub [PubMed PMID: 26540357]
Mohr JP, Parides MK, Stapf C, Moquete E, Moy CS, Overbey JR, Al-Shahi Salman R, Vicaut E, Young WL, Houdart E, Cordonnier C, Stefani MA, Hartmann A, von Kummer R, Biondi A, Berkefeld J, Klijn CJ, Harkness K, Libman R, Barreau X, Moskowitz AJ, international ARUBA investigators. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet (London, England). 2014 Feb 15:383(9917):614-21. doi: 10.1016/S0140-6736(13)62302-8. Epub 2013 Nov 20 [PubMed PMID: 24268105]
Level 1 (high-level) evidenceSpetzler RF, Martin NA. A proposed grading system for arteriovenous malformations. Journal of neurosurgery. 1986 Oct:65(4):476-83 [PubMed PMID: 3760956]
Schramm J, Schaller K, Esche J, Boström A. Microsurgery for cerebral arteriovenous malformations: subgroup outcomes in a consecutive series of 288 cases. Journal of neurosurgery. 2017 Apr:126(4):1056-1063. doi: 10.3171/2016.4.JNS153017. Epub 2016 Jun 10 [PubMed PMID: 27285541]
Level 3 (low-level) evidenceLunsford LD, Niranjan A, Kondziolka D, Sirin S, Flickinger JC. Arteriovenous malformation radiosurgery: a twenty year perspective. Clinical neurosurgery. 2008:55():108-19 [PubMed PMID: 19248675]
Level 3 (low-level) evidenceAdigun R, Goyal A, Hariz A. Systemic Sclerosis (Scleroderma). StatPearls. 2024 Jan:(): [PubMed PMID: 28613625]
Riboldi GM, Samanta D, Asuncion RMD, Frucht S. Ataxia-Telangiectasia. StatPearls. 2024 Jan:(): [PubMed PMID: 30137827]
Blume JE. Generalized essential telangiectasia: a case report and review of the literature. Cutis. 2005 Apr:75(4):223-4 [PubMed PMID: 15916219]
Level 3 (low-level) evidenceRebellato PR, Martins LE, Stella BT, Tokarski MC. A case of benign hereditary telangiectasia without family history. Anais brasileiros de dermatologia. 2017 Jan-Feb:92(1):162-163. doi: 10.1590/abd1806-4841.20174537. Epub [PubMed PMID: 28225986]
Level 3 (low-level) evidenceFarshchian M, Daveluy S. Rosacea. StatPearls. 2024 Jan:(): [PubMed PMID: 32491506]
Costa DL, Moura HH, Rodrigues R, Pineiro-Maceira J, Ramos-E-Silva M. Telangiectasia macularis eruptiva perstans: a rare form of adult mastocytosis. The Journal of clinical and aesthetic dermatology. 2011 Oct:4(10):52-4 [PubMed PMID: 22010057]
Level 3 (low-level) evidenceCallen JP, Wortmann RL. Dermatomyositis. Clinics in dermatology. 2006 Sep-Oct:24(5):363-73 [PubMed PMID: 16966018]
Justiz Vaillant AA, Goyal A, Varacallo M. Systemic Lupus Erythematosus. StatPearls. 2024 Jan:(): [PubMed PMID: 30571026]
Gamboa NT, Joyce EJ, Eli I, Park MS, Taussky P, Schmidt RH, McDonald J, Whitehead KJ, Kalani MYS. Clinical presentation and treatment paradigms of brain arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2018 May:51():22-28. doi: 10.1016/j.jocn.2018.01.019. Epub 2018 Feb 23 [PubMed PMID: 29483005]
Woodall MN, Nakaji P, Spetzler RF. Benefits of Treating Arteriovenous Malformations in Hereditary Hemorrhagic Telangiectasia: A Retrospective Analysis of 14 Patients. World neurosurgery: X. 2019 Jul:3():100029. doi: 10.1016/j.wnsx.2019.100029. Epub 2019 Mar 9 [PubMed PMID: 31225521]
Level 2 (mid-level) evidenceArizmendez NP, Rudmik L, Poetker DM. Intravenous bevacizumab for complications of hereditary hemorrhagic telangiectasia: a review of the literature. International forum of allergy & rhinology. 2015 Nov:5(11):1042-7. doi: 10.1002/alr.21587. Epub 2015 Jul 22 [PubMed PMID: 26202958]
Dupuis-Girod S, Ginon I, Saurin JC, Marion D, Guillot E, Decullier E, Roux A, Carette MF, Gilbert-Dussardier B, Hatron PY, Lacombe P, Lorcerie B, Rivière S, Corre R, Giraud S, Bailly S, Paintaud G, Ternant D, Valette PJ, Plauchu H, Faure F. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012 Mar 7:307(9):948-55. doi: 10.1001/jama.2012.250. Epub [PubMed PMID: 22396517]
Dupuis-Girod S, Ambrun A, Decullier E, Samson G, Roux A, Fargeton AE, Rioufol C, Schwiertz V, Disant F, Chapuis F, Donazzolo Y, Paintaud G, Edery P, Faure F. ELLIPSE Study: a Phase 1 study evaluating the tolerance of bevacizumab nasal spray in the treatment of epistaxis in hereditary hemorrhagic telangiectasia. mAbs. 2014 May-Jun:6(3):794-9. doi: 10.4161/mabs.28025. Epub 2014 Jan 30 [PubMed PMID: 24481211]
Level 1 (high-level) evidenceRiss D, Burian M, Wolf A, Kranebitter V, Kaider A, Arnoldner C. Intranasal submucosal bevacizumab for epistaxis in hereditary hemorrhagic telangiectasia: a double-blind, randomized, placebo-controlled trial. Head & neck. 2015 Jun:37(6):783-7. doi: 10.1002/hed.23655. Epub 2014 Apr 30 [PubMed PMID: 24595923]
Level 1 (high-level) evidenceKarnezis TT, Davidson TM. Treatment of hereditary hemorrhagic telangiectasia with submucosal and topical bevacizumab therapy. The Laryngoscope. 2012 Mar:122(3):495-7. doi: 10.1002/lary.22501. Epub 2011 Dec 6 [PubMed PMID: 22147664]
Stokes P, Rimmer J. Intranasal bevacizumab in the treatment of HHT -related epistaxis: a systematic review. Rhinology. 2018 Mar 1:56(1):3-10. doi: 10.4193/Rhin17.166. Epub [PubMed PMID: 29166422]
Level 1 (high-level) evidenceGuldmann R, Dupret A, Nivoix Y, Schultz P, Debry C. Bevacizumab nasal spray: Noninvasive treatment of epistaxis in patients with Rendu-Osler disease. The Laryngoscope. 2012 May:122(5):953-5. doi: 10.1002/lary.23230. Epub 2012 Mar 23 [PubMed PMID: 22447341]
Dheyauldeen S, Østertun Geirdal A, Osnes T, Vartdal LS, Dollner R. Bevacizumab in hereditary hemorrhagic telangiectasia-associated epistaxis: effectiveness of an injection protocol based on the vascular anatomy of the nose. The Laryngoscope. 2012 Jun:122(6):1210-4. doi: 10.1002/lary.23303. Epub 2012 May 7 [PubMed PMID: 22565282]
Level 3 (low-level) evidenceInvernizzi R, Quaglia F, Klersy C, Pagella F, Ornati F, Chu F, Matti E, Spinozzi G, Plumitallo S, Grignani P, Olivieri C, Bastia R, Bellistri F, Danesino C, Benazzo M, Balduini CL. Efficacy and safety of thalidomide for the treatment of severe recurrent epistaxis in hereditary haemorrhagic telangiectasia: results of a non-randomised, single-centre, phase 2 study. The Lancet. Haematology. 2015 Nov:2(11):e465-73. doi: 10.1016/S2352-3026(15)00195-7. Epub 2015 Oct 27 [PubMed PMID: 26686256]
Level 2 (mid-level) evidenceLebrin F, Srun S, Raymond K, Martin S, van den Brink S, Freitas C, Bréant C, Mathivet T, Larrivée B, Thomas JL, Arthur HM, Westermann CJ, Disch F, Mager JJ, Snijder RJ, Eichmann A, Mummery CL. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nature medicine. 2010 Apr:16(4):420-8. doi: 10.1038/nm.2131. Epub 2010 Apr 4 [PubMed PMID: 20364125]
Level 3 (low-level) evidenceHosman A, Westermann CJ, Snijder R, Disch F, Mummery CL, Mager JJ. Follow-up of Thalidomide treatment in patients with Hereditary Haemorrhagic Telangiectasia. Rhinology. 2015 Dec:53(4):340-4. doi: 10.4193/Rhino14.289. Epub [PubMed PMID: 26735132]
Jameson JJ, Cave DR. Hormonal and antihormonal therapy for epistaxis in hereditary hemorrhagic telangiectasia. The Laryngoscope. 2004 Apr:114(4):705-9 [PubMed PMID: 15064628]
Level 3 (low-level) evidenceSabbà C, Pasculli G, Suppressa P, D'Ovidio F, Lenato GM, Resta F, Assennato G, Guanti G. Life expectancy in patients with hereditary haemorrhagic telangiectasia. QJM : monthly journal of the Association of Physicians. 2006 May:99(5):327-34 [PubMed PMID: 16595564]
Level 2 (mid-level) evidenceTrerotola SO, Pyeritz RE. PAVM embolization: an update. AJR. American journal of roentgenology. 2010 Oct:195(4):837-45. doi: 10.2214/AJR.10.5230. Epub [PubMed PMID: 20858807]
Chamarthy MR, Park H, Sutphin P, Kumar G, Lamus D, Saboo S, Anderson M, Kalva SP. Pulmonary arteriovenous malformations: endovascular therapy. Cardiovascular diagnosis and therapy. 2018 Jun:8(3):338-349. doi: 10.21037/cdt.2017.12.08. Epub [PubMed PMID: 30057880]