Introduction
Heterochromia or heterochromia iridum indicates a difference between the color of the two irises. It can involve the whole iris or only part of the iris (sectoral heterochromia).[1]
It is easier to understand the determinants of iris color with the anatomy of the iris in mind. The iris and the ciliary body constitute the anterior uveal coat. The iris is composed of two anatomical layers that both contain pigment. The iris stroma is the anterior layer, consisting of loose collagenous connective tissue, the sphincter and dilator pupillae muscles, blood vessels, nerves, melanocytes, and other cells. The posterior layer (the iris pigment epithelium) consists of two layers of cuboidal epithelium. The pigment epithelium layer is derived embryologically from the anterior part of the optic cup, and hence neuroectodermal in origin. The iris stroma is mainly mesenchymal in origin. The melanocytes within the iris stroma and the ciliary body are all derived from neurocrest cells.
Another important concept is the amount, type, and location of the pigment present. A type of cell called a melanocyte produces melanin that is deposited in melanosomes. Variation in the number of melanosomes and the amount of pigment in each melanosome present in the anterior iris stroma determines eye color. The type of pigment affects eye color as well. The main two types of pigment are eumelanin (brown to black pigment) and pheomelanin (red to yellow pigment). Eumelanin is present in the iris pigment epithelium, while both eumelanin and pheomelanin are present in the iris stroma. Lipofuscin (yellow in appearance) can accumulate with age and/or ocular disease. Eumelanin is present in the iris, brain, and hair and comes in a brown and black form. Pheomelanin is present in the iris, hair, lips, nipples, glans penis, and vagina and comes in a red and yellow form. The relative presence of the different types of eumelanin and pheomelanin determines both hair and iris color. Both groups of pigment form from L-tyrosine that is converted to L-dopaquinone by tyrosinase. Tyrosinase-related protein-1, in turn, converts L-dopaquinone to eumelanin while the addition of cysteine forms pheomelanin.[2]
Genetics plays an important role in determining eye color, with up to 150 genes involved and two genes, OCA2 and HERC2, on chromosome 15, playing a significant role. OCA2 produces "P protein," which promotes melanosome maturation, and HERC2, in turn, controls OCA2. The inheritance of iris color is largely determined by two genes as well, EYCL1 (also called the gey gene) and EYCL3 (also called the bey2 gene). The gey gene has a green and blue allele, and the bey2 gene has a brown and blue allele. The brown allele is dominant over the green allele, and both are dominant over the blue allele.[3] Since many other genes play a role as well, this occasionally creates unexpected iris color. Congenital heterochromia can be inherited, and autosomal dominant inheritance has been reported.[4] In many cases, however, genetic mosaicism occurs when genetic recombination or a mutation occurs during mitosis, creating an organism with genetically different cells. In albinism, both the iris stroma and the iris pigment epithelium are affected. Oculocutaneous albinism is inherited in an autosomal recessive manner and caused by mutations in tyrosinase, tyrosinase-related protein-1, and OCA2. Ocular albinism is inherited in a sex-linked recessive manner.
Eye color changes from lighter tints to darker during the first year of life, with most changes occurring between 3 and 6 months of age. These changes are dependent on adrenergic innervation. Horner syndrome's lack of adrenergic innervation causes a lighter-colored iris that will have a smaller pupil in dark conditions.[5]
Other structures within the iris and elsewhere may affect the color of the iris as well. First, iris atrophy from conditions such as pigment dispersion syndrome as well as an iridocorneal endothelial syndrome, and iris damage such as caused by surgery or injury can cause heterochromia. Second, anisocoria (different pupil size), which can result from ocular trauma, Adie's pupil, or Horner syndrome, or an abnormal pupil such as is the case with iris coloboma, can create the impression of heterochromia.[6] Neoplastic or hamartomas structures within the iris such as iris naevi and Lisch nodules can cause heterochromia as well. Finally, asymmetrical corneal changes such as band keratopathy, scarring, or edema can create the impression of heterochromia.
Finally, several optical phenomena and light conditions can affect the appearance of the iris. For example, reduced ambient light can cause an amber eye to appear brown. Selective absorption and reflection of other (non-pigment) molecules such as hemoglobin and collagen describe most of the variations in eye color that are not attributed to pigmentation of the iris. However, Rayleigh (causing the blue appearance of the sky) and Tyndall scattering, as well as diffraction, can contribute to eye color as well. Raman scattering and constructive interference (responsible for the coloration of bird and butterfly feathers and brightly colored irises of many animals) do not play a role in human iris coloration.
In addition to the color, human iris tissue may form complex patterns with distinct features. These distinct features can be used for automatic personal identification like fingerprints.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Most cases of heterochromia are sporadic and isolated benign conditions without any clinical significance.
The most important group of conditions with heterochromia are congenital heterochromia which may be the only presentation of congenital Horner syndrome. There may be associated miosis, partial ptosis, and facial anhidrosis. Not all of these signs need to be present to be diagnosed as congenital Horner syndrome. The iris stromal melanocytes derive from neurocrest cells in embryological development. The sympathetic neurons also derive from neurocrest cells. Any disruption of sympathetic innervation of the iris melanocytes due to disruption of the post-ganglionic cervical sympathetic pathways, especially early in life as in congenital Horner syndrome, will result in decreased brown melanin color of one iris and heterochromia. Any syndromic illness associated with impaired neurocrest cells or tissues development and migration may also cause heterochromia, such as Waardenburg syndrome and Parry-Romberg syndrome. Other congenital diseases include ocular melanosis and neurocutaneous syndromes such as Sturge-Weber syndrome (abnormal hyperpigmented iris), oculodermal melanocytosis (nevus of Ota), and neurofibromatosis type I (von Recklinghausen disease, iris hamartoma).[1]
Congenital Causes of Heterochromia
- Benign heterochromia
- Congenital Horner syndrome: with other presentations of miosis, partial ptosis, and/or impaired facial sweating (anhydrosis)[7][5]
- Waardenburg syndrome: hearing loss, heterochromia, lateral displacement of medial canthus, hypertrichosis of medial eyebrows, and prominent broad nasal root[8]
- Parry-Romberg syndrome: progressive facial hemiatrophy[9]
- Sturge-Weber syndrome: facial port-wine stain with meningeal angiomatosis. Patients may present with unilateral neurological deficits, seizures, and a large facial nevus. The ipsilateral eye may have melanocytosis of the uveal coat and retina. Glaucoma is not an uncommon complication.[10]
- Neurofibromatosis I: Lisch nodules in the iris associated with other clinical features. Children with NF1 may also develop optic nerve pathway glioma, which can be detected by simple ophthalmoscopy showing the absence of red reflex in one eye.[11]
- Oculodermal melanocytosis: hamartomatous congenital melanocytosis[12]
- Iridocorneal endothelial (ICE) syndromes[13]
- Iris ectropion syndrome
- Incontinentia pigmenti[14]
Acquired Causes of Heterochromia
- Acquired Horner syndrome
- Other causes of sympathetic heterochromia such as neuroblastoma or paravertebral neurilemmoma[15][16]
- Ocular trauma
- Fuchs heterochromic iridocyclitis[17] (Fuchs heterochromic uveitis)
- foreign body including use of colored contact lens
- Iron deposition: siderosis and hemosiderosis
- Melanocyte infiltration as in diffuse nevus or melanoma
- Local iris tumors
- Certain eyedrops such as prostaglandin analogs[18][19]
Epidemiology
Heterochromia is an uncommon sign. There is no widely reported prevalence.
Pathophysiology
Human iris color exists in a continuum from light blue to dark brown. The most common description uses three shades, including blue, green-hazel, and brown. As discussed above, the most important determining factors for human iris color are the iris stromal texture and stromal melanocytes. The stromal melanocytes are derived from embryological neurocrest cells, which migrate through the uveal tract to the iris during fetal development. Subsequently, the iris stromal melanocytes are also under the trophic influence of the post-ganglionic cervical sympathetic pathway. Consequently, any unilateral lesion affecting the cervical sympathetic (Horner syndrome), either congenital or acquired within the first two years of life, will lead to heterochromia.[5]
Very rarely, longstanding Horner syndrome may also result in heterochromia. Other diseases related to neurocrest cell development and migration may cause heterochromia, such as Waardenburg syndrome and Perry-Romberg syndrome.[8][9]
In addition to diseases primarily involving the iris melanocytes, any local eye or iris disease that causes a unilateral change in colors such as trauma with bleeding, melanoma and other tumors, and Fuchs heterochromic iridocyclitis.[17]
History and Physical
History
- Onset: acute, subacute versus chronic - important in differentiating local eye disease vs. genetic causes. Progressive facial hemiatrophy is typical for Parry-Romberg syndrome.
- Any local eye symptoms such as pain, redness, and visual disturbances suggest local eye diseases.
- History of trauma.
- History of use of eye drops, especially used in one eye, causing discoloration of that eye such as latanoprost and other prostaglandin analog eyedrops.[18]
- Family history of neurofibromatosis type I
Physical
- Most important: Review old photos, especially childhood pictures. Look for the presence of heterochromia in the past and signs of Horner syndrome such as miosis and partial ptosis.
- Look for other signs of Horner syndrome - miosis and ptosis.
- Look for signs of specific syndromes such as cutaneous neurofibromata, facial hemiatrophy, facial port-wine stains, hearing loss, and broad facial features.
- Careful exam of the eye, especially the iris, anterior chamber, pupil size and responses, corneal conditions, and intraocular pressure measurement.[12]
Evaluation
Eye examination by an ophthalmologist with a slit lamp examination. Clinical exam, especially to look for other signs of Horner syndrome.[16]
Clinicians can use urinary catecholamines, VMA, etc., for excessive sympathetic activities in children to rule out underlying neuroblastoma and other neurocrest derived tumors.
CT chest and abdomen to causes of Horner syndrome or possible neuroblastoma or other tumors in children and infants.[20] CT angiogram and CT skull base for hypoplasia of the internal carotid artery as proof of congenital Horner syndrome, or evidence of carotid dissection in chronic acquired Horner syndrome.
Most of the time, with obviously isolated heterochromia and no other abnormal findings on physical exam, the diagnosis will be benign heterochromia. However, no further evaluation plus reassurance is adequate for the management of the patient.
Treatment / Management
Heterochromia is a sign and not by itself a disease. There is no specific treatment of heterochromia by itself. Treatment is directed at the underlying cause, including underlying eye diseases. If the heterochromia is associated with other signs of Horner syndrome such as miosis and partial ptosis, the most common cause is congenital Horner syndrome which does not require any treatment. On the other hand, congenital or early childhood onset Horner syndrome may result from an underlying tumor of the sympathetic chain, especially neuroblastoma which will need urgent workup, diagnosis, and treatment.[21] Heterochromia caused by an acquired Horner syndrome, especially in adults, signifies a chronic and longstanding illness. The underlying cause is very unlikely to be a malignancy.
Differential Diagnosis
Heterochromia is a clinical sign. The differential diagnosis will include all the causes listed above. The most important differential diagnoses to consider are:
Prognosis
The prognosis of heterochromia depends on the underlying cause. In infants and children, it is important to rule out underlying neoplasia of the sympathetic chain, especially neuroblastoma which carries a poor prognosis when diagnosed and treated late.[22] Fuchs heterochromic iridocyclitis may threaten vision due to complications of glaucoma. An iris melanoma may be malignant. The most common cause is benign heterochromia with no clinical significance. The other significant group of causes is congenital Horner syndrome which usually has a good prognosis.[16]
Complications
Heterochromia is a clinical sign, and there is no health complication due to this sign per se.
Consultations
Ophthalmological consultation is indicated for most causes of local eye diseases causing heterochromia. Oncology consultation is often necessary with suspected underlying neuroblastoma in children.[23]
Pearls and Other Issues
It is most important to look at old pictures of the patient, especially childhood pictures. The presence of heterochromia since early childhood usually indicates benign heterochromia or congenital Horner syndrome.[16] No further workup is indicated, and the patient should be reassured.
Enhancing Healthcare Team Outcomes
Heterochromia is a rare yet fascinating sign found in some patients, and it may cause unnecessary worries for the patients and families. These patients are usually first seen by their primary care doctors. Interprofessional care and communications between the primary care physician, ophthalmologist, and neurologist will be most effective in determining the cause of the patient's heterochromia and appropriate management or reassurance.
Making the final common diagnosis of benign heterochromia or congenital Horner syndrome by the healthcare team also helps the patient and family to understand the benign nature of the problem and reassurance about it being a non-inherited problem.
Media
(Click Image to Enlarge)
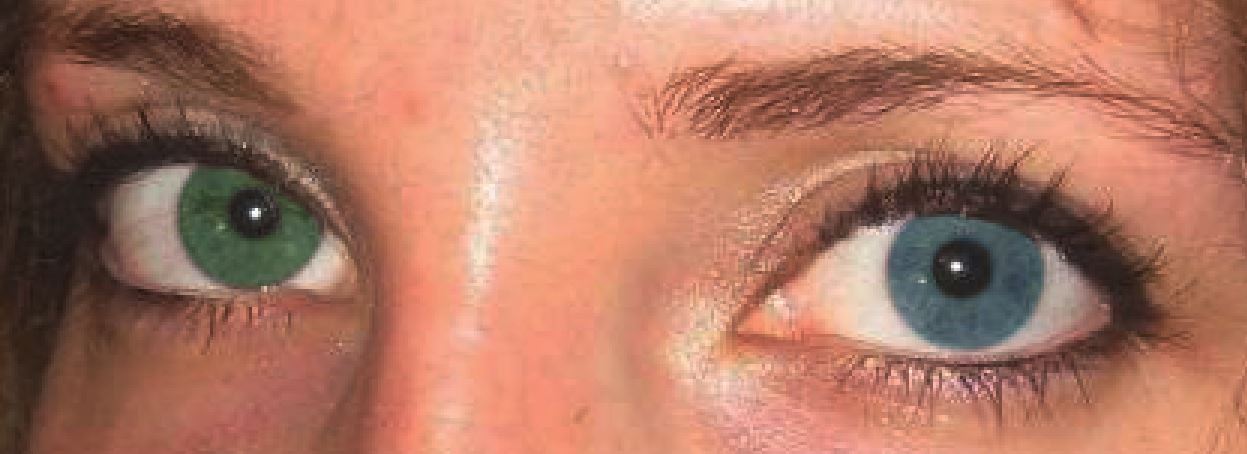
Example of heterochromia. Created by wiki user Deanern1, used under Attribution-ShareAlike 3.0 Unported (CC BY-SA 3.0), https://creativecommons.org/licenses/by-sa/3.0/
References
Ur Rehman H, Heterochromia. CMAJ : Canadian Medical Association journal = journal de l'Association medicale canadienne. 2008 Aug 26; [PubMed PMID: 18725617]
Level 3 (low-level) evidenceProta G,Hu DN,Vincensi MR,McCormick SA,Napolitano A, Characterization of melanins in human irides and cultured uveal melanocytes from eyes of different colors. Experimental eye research. 1998 Sep [PubMed PMID: 9778410]
Imesch PD,Wallow IH,Albert DM, The color of the human eye: a review of morphologic correlates and of some conditions that affect iridial pigmentation. Survey of ophthalmology. 1997 Feb [PubMed PMID: 9154287]
Level 3 (low-level) evidenceTomar M,Dhiman R,Sharma G,Yadav N, Artistic Iris: A Case of Congenital Sectoral Heterochromia Iridis. Journal of ophthalmic & vision research. 2018 Jul-Sep [PubMed PMID: 30090196]
Level 3 (low-level) evidenceRenard D,Jeanjean L,Labauge P, Heterochromia Iridis in congenital Horner's syndrome. European neurology. 2010; [PubMed PMID: 20375515]
Level 3 (low-level) evidenceDiagne JP,Sow AS,Ka AM,Wane AM,Ndoye Roth PA,Ba EA,De Medeiros ME,Ndiaye JM,Diallo HM,Kane H,Sow S,Nguer M,Sy EM,Ndiaye PA, [Rare causes of childhood leukocoria]. Journal francais d'ophtalmologie. 2017 Oct [PubMed PMID: 28893456]
Deprez FC,Coulier J,Rommel D,Boschi A, Congenital horner syndrome with heterochromia iridis associated with ipsilateral internal carotid artery hypoplasia. Journal of clinical neurology (Seoul, Korea). 2015 Apr; [PubMed PMID: 25749818]
Liu Y,Pan H,Wang J,Yao Q,Lin M,Ma B,Li J, Ophthalmological features and treatments in five cases of Waardenburg syndrome. Experimental and therapeutic medicine. 2020 Oct; [PubMed PMID: 32855674]
Level 3 (low-level) evidenceArif T,Fatima R,Sami M, Parry-Romberg syndrome: a mini review. Acta dermatovenerologica Alpina, Pannonica, et Adriatica. 2020 Dec; [PubMed PMID: 33348939]
Hassanpour K,Nourinia R,Gerami E,Mahmoudi G,Esfandiari H, Ocular Manifestations of the Sturge-Weber Syndrome. Journal of ophthalmic [PubMed PMID: 34394871]
Pinti E,Nemeth K,Staub K,Lengyel A,Fekete G,Haltrich I, Diagnostic difficulties and possibilities of NF1-like syndromes in childhood. BMC pediatrics. 2021 Jul 29; [PubMed PMID: 34325699]
Sarker BK,Malek MA,Abdullahi S,Iftekhar S,Sakeb N,Mahatma M,Sarkar MK, Ophthalmic associations of oculodermal melanocytosis in a tertiary eye hospital in South Asia. Therapeutic advances in ophthalmology. 2021 Jan-Dec; [PubMed PMID: 33644684]
Level 3 (low-level) evidenceChaurasia S,Choudhari NS,Mohamed A, Clinical and Specular microscopy characteristics and corelation in Iridocorneal endothelial syndrome without corneal edema. Seminars in ophthalmology. 2021 Oct 3 [PubMed PMID: 33750265]
Cohen BA, Incontinentia pigmenti. Neurologic clinics. 1987 Aug [PubMed PMID: 3306331]
Wallis DH,Granet DB,Levi L, When the darker eye has the smaller pupil. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2003 Jun; [PubMed PMID: 12825064]
Level 3 (low-level) evidencePérez-Torres-Lobato MR,De Las Morenas-Iglesias J,Llempén-López M,Gómez-Millán-Ruiz P,Márquez-Vega C,Espiñeira-Periñán MÁ,Coronel-Rodríguez C,Franco-Ruedas C,Balboa-Huguet B,Sánchez-Vicente JL, Paediatric Horner syndrome. A case series of 14 patients in a tertiary hospital. Archivos de la Sociedad Espanola de Oftalmologia. 2021 Jul; [PubMed PMID: 34217473]
Level 2 (mid-level) evidenceMoshirfar M,Villarreal A,Ronquillo Y, Fuchs Uveitis Syndrome StatPearls. 2021 Jan; [PubMed PMID: 32644574]
Stjernschantz JW,Albert DM,Hu DN,Drago F,Wistrand PJ, Mechanism and clinical significance of prostaglandin-induced iris pigmentation. Survey of ophthalmology. 2002 Aug; [PubMed PMID: 12204714]
Level 3 (low-level) evidenceDoyle E,Liu C, A case of acquired iris depigmentation as a possible complication of levobunolol eye drops. The British journal of ophthalmology. 1999 Dec [PubMed PMID: 10660314]
Level 3 (low-level) evidenceMehta K,Haller JO,Legasto AC, Imaging neuroblastoma in children. Critical reviews in computed tomography. 2003; [PubMed PMID: 12627783]
Abramson SJ,Berdon WE,Ruzal-Shapiro C,Stolar C,Garvin J, Cervical neuroblastoma in eleven infants--a tumor with favorable prognosis. Clinical and radiologic (US, CT, MRI) findings. Pediatric radiology. 1993 [PubMed PMID: 8414748]
Irwin MS,Naranjo A,Zhang FF,Cohn SL,London WB,Gastier-Foster JM,Ramirez NC,Pfau R,Reshmi S,Wagner E,Nuchtern J,Asgharzadeh S,Shimada H,Maris JM,Bagatell R,Park JR,Hogarty MD, Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2021 Jul 28; [PubMed PMID: 34319759]
Grant CN,Rhee D,Tracy ET,Aldrink JH,Baertschiger RM,Lautz TB,Glick RD,Rodeberg DA,Ehrlich PF,Christison-Lagay E, Pediatric solid tumors and associated cancer predisposition syndromes: Workup, management, and surveillance. A summary from the APSA cancer committee. Journal of pediatric surgery. 2021 Aug 24 [PubMed PMID: 34503817]