
Introduction
Retinal dystrophies or inherited retinal diseases (IRD) are a group of degenerative disorders of the retina with clinical and genetic heterogeneity.[1] Common presentations include dimness of vision, color blindness, night blindness, peripheral vision abnormalities, and subsequent progression to complete blindness in progressive conditions. Multiple causative gene defects have been identified with more than 270 genes linked to various retinal dystrophy types and clinical presentations varying even among family members with the same mutation.[2][3]
Diagnosis often involves genetic analysis, retinal imaging, and electrophysiological tests. Supportive care, genetic counseling, and psychological support play essential roles in managing these progressive disorders. While there is no cure, treatments like gene therapy approved by the United States Food and Drug Administration (FDA) for specific conditions like Leber congenital amaurosis, retinal prostheses, and low-vision aids aim to enhance patients' quality of life.[4]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Rods and cone photoreceptors are the central cellular units responsible for visual phototransduction.[5] Phototransduction is a process by which light signals are converted into action potentials within the retina, facilitating the brain's image perception. During this process, light-sensitive pigments are generated and recycled. Etiologies of retinal dystrophies include abnormalities in photoreceptors and defects in phototransduction.
Depending on the type of photoreceptors affected, retinal dystrophies can be subdivided into rod-dominated diseases, cone-dominated diseases, and generalized forms involving both rods and cones. Cases may be syndromic, nonsyndromic, sporadic, or familial. In familial cases, inheritance patterns could be autosomal dominant, autosomal recessive, or X-linked. Syndromic cases have clinical features that extend to multisystemic involvement rather than nonsyndromic varieties, which affect only the retina.[6]
The Iowa classification of IRD has 3 clinical types.[7] The photoreceptor disease type is the most common (64.7%). Retinitis pigmentosa is the major disease in this category. The second type is macular diseases, which include autosomal recessive Stargardt disease, pattern dystrophy, and Best disease.[8] The last type is "third branch disease," which includes other IRDs, including retinoschisis, optic neuropathy, and choroidopathies, such as gyrate atrophy of the choroid and retina.[7][9] Approximately 90% of patients with retinal dystrophies receive one of the following diagnoses, also called the "Big 14" according to the Iowa classification:[7]
Epidemiology
The exact incidence of retinal dystrophies is unknown; the most common retinal dystrophy, retinitis pigmentosa, affects around 1 in 5000 individuals worldwide.[13] Other dystrophies, such as achromatopsia, are rarer, with an incidence of 1:30000.[14] Most of these dystrophies affect children and working-age young adults, adding to the socioeconomic burden.
The most commonly mutated genes that cause retinal dystrophies include:
- ABCA4: The most common gene associated with IRDs, accounting for around 12.9% of cases
- USH2A: The second most common gene, accounting for around 6.8% of cases
- RPGR: The third most common gene, accounting for around 2.7% of cases [15][16]
Other genes causing retinal dystrophies include EYS, RHO, PRPH2, BEST1, RS1, RP1, CHM, CRB1, PRPF31, MY07A, and OPA1.[16] The so-called "North Carolina macular dystrophy" appears to be a misnomer as it has been reported worldwide, including in other parts of the United States, the United Kingdom, Europe, Australia, China, and Turkey.[17][18][19] The Island of the Colorblind is the nickname of Pingelap, a Micronesian atoll in the South Pacific.[20] Some families within that area have complete achromatopsia (ie, rod monochromacy or Pingelapese blindness).[14]
Pathophysiology
Mutations in various genes cause retinal dystrophies. These mutations can be inherited in an autosomal dominant, autosomal recessive, X-linked, or mitochondrial manner. The specific gene mutations determine the type of IRD, such as retinitis pigmentosa, Leber congenital amaurosis, or Stargardt disease.
The primary pathology in retinal dystrophies involves the dysfunction and death of photoreceptor cells (rods and cones) and the retinal pigment epithelium (RPE), including:
- Protein misfolding and aggregation: Mutations often produce misfolded proteins that aggregate within retinal cells, causing cellular stress and apoptosis.
- Defective phototransduction: Mutations in genes involved in the phototransduction cascade can impair light conversion into electrical signals, leading to photoreceptor cell death.
- Oxidative stress: Accumulating reactive oxygen species due to mitochondria dysfunction or impaired antioxidant defenses contributes to cellular damage and apoptosis.
- Inflammation: Chronic inflammation and microglia activation in the retina can exacerbate photoreceptor degeneration.
- Impaired retinal metabolism: Mutations affecting metabolic pathways in the retina can lead to energy deficits and cell death.[21]
Dysfunction of photoreceptors' outer segment is an important aspect of retinal dystrophies.[22][21] The crucial functions of these affected outer segments in various retinal dystrophies include:
- Phototransduction: The mutations affecting phototransduction may involve multiple genes, including rhodopsin (RHO), PDE6, and cyclic nucleotide-gated (CNG) channels.[23]
- Disc morphogenesis: A crucial gene for disc morphogenesis includes PRPH2.[24]
- Ciliary formation or trafficking: Major genes controlling cilia formation include USH2A (affected in Usher syndrome), RPGR, CEP290, and BBS1-22 (affected in Bardet-Biedl syndrome). The syndromes associated with ciliopathy and retinal dystrophies include Usher, Bardet-Biedl, Joubert, Senior–Løken, and Alstrom syndromes.[25][26][21]
Dysfunctions of RPE can affect the visual cycle, phagocytosis with a resultant turnover of photoreceptor outer segments, lipid homeostasis, ionic regulation, and pigment formation.[27] The most important genes affecting these include ABCA4, causing Stargardt disease, and VMD2, which encodes BEST1 or bestrophin; VMD2 mutations cause Best disease.[28][8] Affection of development, homeostasis, retina metabolism, and choroid can also cause retinal dystrophies.[21] The other changes involved in retinal dystrophies include cell adhesion, changes in the basement membrane and extracellular matrix, energy metabolism of the retina and RPE, peroxisome activity, autophagy, iron regulation, cell signaling, leucocyte activation, and activation of proinflammatory pathways.[21]
Histopathology
The anatomical areas involved in respective types of retinal dystrophy include:
- Photoreceptor disease: cones and rods
- Macular disease: the area between photoreceptors and retinal pigment epithelium (RPE)
- Third branch disease: superficial retina or deeper choroid, depending on the disease [7]
Retinitis pigmentosa is characterized histopathologically by progressive degeneration of the retinal photoreceptors, especially the rods, followed by cones.[29] The rods die due to various mutations and mechanisms, including apoptosis.[30] After the death of rod cells, there is a high oxygen level in the outer retina, and the remaining cones undergo progressive oxidative damage.[30] This is accompanied by reactive gliosis, loss of outer retinal layers, and migration of pigment-laden cells from the RPE and macrophages into the neural retina, especially around vessels.[31] The common findings are progressive thinning of the retinal layers, especially the outer nuclear layer, and eventual atrophy of the RPE and choriocapillaris.[32] In advanced stages, optic nerve head pallor and vascular attenuation become pronounced.[31] Please see StatPearls' companion resource "Retinitis Pigmentosa," for further information.
Cone dystrophy is histopathologically marked by selective degeneration of cone photoreceptors, initially affecting the central macula, with sparing of rod photoreceptors until later stages. This leads to thinning of the outer nuclear layer, loss of cone outer segments, and progressive atrophy of the retinal pigment epithelium (RPE), primarily in the macular region.[33] In cone-rod dystrophy, cones are affected first, followed by the involvement of the rods later, which is opposite to retinitis pigmentosa (ie, rod-cone dystrophy).[34] Macula and visual acuity are involved early in cone-rod dystrophy.[34]
Stargardt disease is histopathologically marked by lipofuscin accumulation within the RPE due to dysfunctional ABCA4 gene expression. This impairs the clearance of vitamin A and its metabolic byproducts. This leads to progressive degeneration of the RPE and secondary photoreceptor loss, particularly in the macular region. Histologic examination reveals atrophy of the outer retinal layers, including the photoreceptor outer segments. Cones are more severely damaged than rods.[35] Müller cells may show reactive hypertrophy.[36] The appearance of yellowish, pisciform flecks in ophthalmoscopy is due to abnormal lipofuscin deposits. Over time, the macular region with geographic atrophy of the RPE and choriocapillaris has profound thinning (see Image. Stargardt Disease).[8] Please see StatPearls' companion reference, "Stargardt Disease," for further information.
Best disease, or Best vitelliform macular dystrophy, is characterized histopathologically by the accumulation of lipofuscin-like material within the RPE, leading to the formation of the characteristic vitelliform lesion.[37] This material disrupts normal RPE function, resulting in the degeneration of the overlying photoreceptors over time, particularly in the macula.[38] However, these patients usually have good vision despite the prominent fundus appearance. As the disease progresses, the vitelliform material can rupture, leading to a pseudohypopyon stage, followed by atrophy of the RPE and outer retinal layers, resulting in fibrosis and choroidal neovascularization in advanced stages. Histologic findings in the late stage include RPE thinning, loss of photoreceptor outer segments, and subretinal fibrosis.[28] The histopathology of other dystrophies varies. Please see StatPearls' companion reference, "Best Disease," for further information.
History and Physical
The functions of rods include scotopic vision (night vision or low-light vision) and peripheral vision. Rod dysfunction results in night blindness (nyctalopia), delayed dark adaptation, and peripheral visual field constriction. Cone dysfunction results in poor central vision, central scotoma, color blindness, photophobia, nystagmus, and day blindness (hemeralopia).[39]
The primary clinical history that should be obtained, according to the University of Iowa Inherited Eye Disease history form, from a suspected patient with retinal dystrophies includes:
- Patient age at symptom onset
- Initial symptoms
- Initial diagnosis
- Previous diagnoses [7]
- Syndromic features: Usher syndrome is characterized by retinitis pigmentosa, hearing loss, and balance abnormality; Bardet-Biedl syndrome is associated with retinitis pigmentosa, polydactyly, short height, obesity, hypogonadism, and renal involvement.
- Use of long-term medications that can impact vision or the eye: Long-term use of drugs, including thioridazine, can cause a retinitis pigmentosa-like fundus picture. Chloroquine and hydroxychloroquine may cause bull's eye maculopathy and simulate cone dystrophy. Pentosan polysulfate can cause macular changes similar to Stargardt disease. However, the lesions usually do not spare the area around the optic disc.[40] Didanosine can cause fundus appearance similar to gyrate atrophy of the retina and retinal pigment epithelium.[41]
- Any history of cancer or autoimmune disease: Autoimmune retinopathy (AIR) may cause recent-onset nyctalopia in adults or older individuals. AIR may be paraneoplastic or nonparaneoplastic. Paraneoplastic AIR includes cancer-associated retinopathy (CAR), melanoma-associated retinopathy (MAR), bilateral diffuse uveal melanocytic proliferation (BDUMP), and paraneoplastic optic neuropathy (PON).[42]
- Any relevant family history: The family history helps create a pedigree chart that gives insight into the inheritance pattern.
- History of driving: Patients with retinitis pigmentosa may have a constricted visual field, though their central vision may be excellent. Due to the constricted visual field, the patient may not meet the minimum requirement for driving in various countries.
- Refraction of both eyes: Usually, retinal dystrophies, including retinitis pigmentosa, cone-rod dystrophy, gyrate atrophy of the retina, and RPE, are associated with myopia. Hyperopia is noted in retinal dystrophies, including Leber congenital amaurosis and Best disease.[28]
- Color vision test: Color vision is affected by cone dysfunction.
Additionally, to differentiate between rod-selective or cone-selective diseases, specific clinical features support the correct diagnosis, including:
- The clinical triad of cone dysfunction includes photo-dysphoria, dyschromatopsia, and poor central vision with or without nystagmus. Fundus findings of cone dystrophy affect the fovea and macula first, and the optic disc and retinal vessels are usually unremarkable. Also, bony spicule pigmentation at the mid-peripheral fundus is not seen in typical cone dysfunction.
- Rod dysfunction is characterized by poor dark adaptation, poor vision at night (nyctalopia), and loss of peripheral vision. On the fundus, the rod dysfunction causes attenuation of retinal arteries, optic disc pallor, and bony spicule pigmentation starting at the mid-periphery of the fundus.
- Preference for a too-dark (cone dystrophy) or a too-bright room (rod dystrophy)
- Difficulty in a movie theatre or similar dim environments (rod dysfunction)
- Ability to see the general outlines of objects under ambient lighting conditions (eg, after waking up at night and in the presence of night lights)
- Disorientation at night during a camping trip, when others can find their way easily (rod dysfunction)
- Avoidance of bright light or being uncomfortable on a sunny day (cone dystrophy)
- Worse color vision than that of peers (cone dystrophy) [7]
Photoreceptor Diseases
Rod dystrophies
Rod-dominated dystrophies include rod and rod-cone dystrophies, in which rod photoreceptors are predominantly or the first affected. These include retinitis pigmentosa and congenital stational night blindness (CSNB).
Retinitis pigmentosa
Retinitis pigmentosa is the most commonly seen retinal dystrophy. The prevalence is around 1 in 5000.[10] Retinitis pigmentosa is a progressive rod-cone disease with rods affected first and has a high level of clinical and genetic heterogeneity. The age of presentation and the prognosis depends on the type of inheritance. Like other forms of retinal dystrophies, it may be sporadic or inherited in an autosomal dominant (AD), autosomal recessive (AR), or X-linked recessive (XLR) pattern. AD is the least severe form, and XLR is the most severe. Nonsyndromic and syndromic forms are reported. The most common genes associated with retinitis pigmentosa include:
- RHO in autosomal dominant cases
- USH2A in autosomal recessive cases
- RPGR in X-linked recessive cases [43]
Nyctalopia is a constant feature, although it is not pathognomic of retinitis pigmentosa. Most cases do not report night vision problems until the ocular disease is advanced. Patients also notice an insidious progressive loss of peripheral field of vision. In most cases, the inferior retina is affected first, denoting light exposure's possible role. Hence, superior field losses are seen early. Visual field loss and night vision problems make these patients prone to accidents, especially at night. In typical retinitis pigmentosa, the rate of progression of visual field loss is usually slow. Usually, the patient does not notice these changes until it reaches the stage of tunnel vision when the patient becomes acutely aware of the changes. Central vision can be affected earlier due to secondary changes, including cystoid macular edema, epiretinal membrane, vitreomacula traction, macular hole, or the development of RPE defects in the macular or fovea.[44] In most cases, color vision remains unaffected until the later stages of the disease.
Fundus appearance of retinitis pigmentosa classically includes a triad of retinal vessel attenuation, waxy pallor of the optic disc, and bone spicule intraretinal pigmentation (see Image. Retinitis Pigmentosa). Patients with very early retinitis pigmentosa without fundus pigmentary abnormalities are termed retinitis pigmentosa sine pigmento. However, this is no longer considered a subtype of retinitis pigmentosa as it is a stage of retinitis pigmentosa in some patients.[45] Such patients who do not show typical bony spicule pigments usually show changes in fundus autofluorescence and visual field changes.[46] Fine dust-like pigment cells are noted in the vitreous cavity released from the degeneration of RPE.
Phenotypic variants based on retinal involvement are sectoral retinitis pigmentosa, pericentral retinitis pigmentosa, and unilateral or extremely asymmetric retinitis pigmentosa. Sectoral retinitis pigmentosa is characterized by pigmentary changes limited to 1 or 2 quadrants with limited visual field changes, good electroretinogram (ERG) responses, and minimal progression with time. Sector retinitis pigmentosa can be autosomal dominant or recessive. Sporadic cases are common and may result from nongenetic causes of retinal degeneration. The pericentral variant shows field loss between 5 and 15 degrees from fixation. The areas of field deficit enlarge and coalesce over time, causing early encroachment on the central field of vision and more significant disability. Unilateral retinitis pigmentosa is typically an acquired condition with diffuse unilateral subacute neuroretinitis (DUSN) as an important differential diagnosis. However, unilateral retinitis pigmentosa may have a genetic association.[47]
Congenital stationary night blindness
CSNB is a nonprogressive form of night blindness that has been present since birth. Various inheritance patterns, including autosomal dominant, autosomal recessive, and X-linked, are now recognized. The mutations usually cause defects in the phototransduction or postphototransduction transmission. CSNB is further subcategorized as CSNB with normal fundus and CSNB with abnormal fundus. Cases of CSNB with normal fundus present may be inherited in autosomal recessive, autosomal dominant, and X-linked recessive modes.
CSNB with abnormal fundi includes 2 entities: Oguchi disease and fundus albipunctatus. In Oguchi disease, the Mizuo-Nakamura phenomenon is classically observed.[48] In this variant, a golden sheen over the retina is noted, and an unusually dark fovea is observed when exposed to light. However, the retina appears normal after prolonged dark adaptation. Color vision and visual acuity are standard in these cases. Histopathological studies suggest the presence of an abnormal layer between the outer segments of photoreceptors and RPE.[49] Fundus albipunctatus is another type of CSNB with yellow-white dots over the fundus. In most cases, these dots are found incidentally on routine eye checkups. Genes involved in fundus albipunctatus include RDH5 and RLBP1.[50]
Cone disorders
Cone-dominated diseases can be further subdivided into early-onset forms with no progression and late-onset forms that are usually progressive. The progressive forms include cone-dominated dystrophies and cone dystrophies. The stationary forms include achromatopsia and blue cone monochromatism.
Achromatopsia
Achromatopsia is an autosomal recessive condition in which patients present with poor visual acuity, photosensitivity, and poor color discrimination since birth. Photosensitivity is due to poor visual acuity in bright lights instead of light intolerance. Photosensitivity is subtyped as complete and incomplete achromatopsia.[14] Cases with complete or total achromatopsia (see Table. Comparison of Rod Monochromatism and Blue-Cone Monochromatism), also known as rod monochromats, usually have visual acuity of less than 20/200.
Incomplete or atypical forms retain a visual acuity between 20/60 and 20/200. Color vision is completely absent in complete achromatopsia cases. Nystagmus may be present from 2 to 3 months of age but usually improves with time. Photophobia also improves with time. Fundus examination is generally normal in these cases; however, some cases may have a granular appearance of the macula or temporal optic disc pallor. Visual field testing may reveal central scotoma; peripheral fields are usually normal or mildly constricted. Characteristically, these are nonprogressive changes.
Cone monochromatism
Cone monochromatism is an X-linked recessive congenital disorder where 2 of the 3 cone systems are absent or significantly affected. The most common variety is blue cone monochromatism, in which both red and green cone systems are absent. Visual acuity in affected individuals ranges from 20/60 to 20/200. Clinical signs and symptoms resemble achromatopsia cases. Typically, patients can perceive blue color.[51]
Table. Comparison of Rod Monochromatism and Blue-Cone Monochromatism
| Rod Monochromatic (complete achromatopsia, absent cone function, complete color-blindness, day blindness, Pingelapese blindness) | Blue-Cone Monochromatism (blue cone monochromacy, X-linked incomplete achromatopsia, S-cone monochromacy, or atypical X-linked achromatopsia) | |
| Incidence |
|
|
| Stability |
|
|
| Etiology |
|
|
| Gender |
|
|
| Visual acuity |
|
|
| Nystagmus |
|
|
| Color Vision |
|
|
| Other Features |
|
|
| Intolerance to Light |
|
|
| Inheritance |
|
|
| Genes |
|
|
| Fundus |
|
|
| Fundus Autofluorescence |
|
|
| Optical Coherence Tomography of the Macula |
|
|
| Electroretinogram |
|
|
| Important Geographic Associations |
|
Cone-rod dystrophy
Cone-rod dystrophy (CRD) is usually misdiagnosed as retinitis pigmentosa with more involvement of cones than rods. Patients present with early loss of vision and color vision abnormalities due to cone dysfunction with subsequent peripheral field constriction due to rod dysfunction. The fundus examination reveals macular pigmentation and atrophy in the early stages, followed by peripheral bone spicule pigmentation in advanced cases. Often, mid-periphery is affected later in the course of the disease. The diagnosis of CRD is essentially based on ERG changes, which show cone affliction more than rods.
Generalized retinal dystrophies
This category of photoreceptor diseases includes disorders like LCA. LCA is a group of disorders due to a mutation in at least 17 genes, all presenting with severe visual impairment or blindness from infancy and extinguished ERG (dysfunction of both rods and cones). Nystagmus is usually present, and the onset is before 4th birthday. Most patients show either a normal fundus appearance or subtle RPE changes and retinal vascular attenuation. Most patients have hyperopia. Eye rubbing, also known as the oculo-digital sign, is a common association. Keratoconus is seen in 29% of cases.[59][60] Enophthalmos may be noted due to the atrophy of orbital fat as a consequence of eye rubbing, as well as other genetic factors. Complicated cases have systemic features. Cases of deafness, renal anomalies, hepatic dysfunction, and skeletal abnormalities have been reported. The most common inheritance pattern is autosomal recessive. Patients with RPE65 or CRB1 gene mutations have progressive vision loss with age, with a slightly better prognosis in those with onset after infancy.[61] Clinically, the LCA spectrum has been subdivided into the following descriptive terms according to some authors:
- No educationally useful vision (eg, LCA); CEP290 is the most common gene involved.
- Educationally useful vision initially, but legally blind due to poor central vision or constricted visual field before the tenth birthday (eg, SECORD).
- Not legally blind at 10 years of age (eg, ECORD) [7]
Syndromic photoreceptor disease
Usher and Bardet-Biedl syndrome are the most common syndromes associated with photoreceptor disease. Usher syndrome, or Hallgren syndrome, is a rare genetic condition characterized by progressive vision and hearing loss. Usher syndrome has been classified into 3 major subtypes: I, II, and III, according to the genes responsible and the onset and severity of the signs and symptoms. Usher syndrome type I is the most severe subtype, characterized by marked congenital hearing loss, retinitis pigmentosa, and absent vestibular function. Usher syndrome type II is less severe. Patients with this subtype have moderate-to-severe congenital hearing loss, retinitis pigmentosa, and normal vestibular function. Usher syndrome type III involves progressive hearing loss, retinitis pigmentosa, and varying degrees of vestibular dysfunction. Onset for type III is typically within the second to fourth decades of life. These patients also tend to have better vision than the other subtypes.
The most common genes associated with Usher syndrome include:
- MYO7A in type 1, accounting for more than 50% of cases
- USH2A in type 2, accounting for approximately 80% of cases
- CLRN1 in type 3, the most common gene associated with this type of Usher syndrome [62]
Bardet-Biedl syndrome (BBS) is a rare, autosomal recessive genetic disorder that can lead to dysfunction of multiple organ systems, including the kidneys, genitalia, brain, and eye. BBS is caused by pathogenic mutations in genes encoding proteins involved in the function of nonmotile primary cilia. The most commonly mutated gene is BBS1 (23% of cases), followed by BBS10 (15%) and BBS2 (10%). Fundus examination typically shows pigmentary degeneration with early macular atrophy and vascular attenuation. Pigmentary changes are seen (see Image. Pigmentary Retinopathy). A majority of patients also exhibit early obesity, polydactyly, and intellectual impairment.[11]
Macular Diseases
Stargardt disease
Stargardt disease, or fundus flafvimaculatus, is the most common childhood recessively inherited macular dystrophy. The condition has a genetic basis due to mutations in the ABCA4 gene on chromosome 1. This results in accumulating visual cycle kinetics-derived byproducts in the RPE with secondary photoreceptor dysfunction and death. Stargardt patients may be asymptomatic but most commonly present with bilateral central visual loss, photophobia, color vision abnormalities, central scotomas, and slow dark adaptation. The fundus may appear normal, considering the severity of vision loss. The fundus features include vermillion fundus, macular atrophy, beaten bronze or metallic sheen at the fovea, and pisciform subretinal flecks at the posterior pole surrounding the fovea and the optic disc typically sparing the peripapillary region (see Image. Stargardt Disease).[8] Fluorescein angiography usually shows dark choroid.[63] Mutation of the ABCA4 gene can cause multiple phenotypes.
Best disease
Best disease (best vitelliform macular dystrophy) is a rare autosomal dominant disorder due to BEST1 (or VMD2, bestrophin-1, or TU15B) gene mutation with incomplete penetrance and variable expression. Yellow vitelliform material is seen subretinally.[28] Typically, an egg yolk-like subretinal lesion is noted at the macula of both eyes with good vision (see Image. Imaging in Best Disease).[64] Electrooculogram is typically affected with the Arden ratio of less than 1.5. Mutation of the VMD2 gene can cause various phenotypes, including:
- Best vitelliform macular dystrophy
- Autosomal recessive dystrophinopathy
- Retinitis pigmentosa-50
- Retinitis pigmentosa (concentric)
- Vitreoretinochoroidopathy
Pattern dystrophies
Pattern dystrophies are a group of autosomal dominant macular diseases characterized by various patterns of pigment deposition within the macula. However, most cases of adult-onset vitelliform macular dystrophy are sporadic.[65] The primary layer of the retina affected is the RPE. RPE is responsible for removing and recycling waste within the retina. In various pattern dystrophies, this waste accumulates in the form of lipofuscin. These findings were initially attributed to mutations in the PRPH2 gene (also known as RDS), which provides instructions for making a protein called peripherin-2. Other genes include ABCA4, VMD2, and IMPG1. Considerable genetic and phenotypic heterogeneity can be present.[66] Based on the pattern of the pigment distribution, they have been classified as:
- Adult-onset foveomacular vitelliform dystrophy or adult-onset vitelliform macular dystrophy
- Sjögren reticular pigment epithelial dystrophy
- Butterfly-shaped pigment dystrophy
- Multifocal pattern dystrophy simulating Stargardt disease
- Fundus pulverulentus, pattern dystrophy appearance may transform into another type of pattern dystrophy with time [67]
Third Branch Disorders
Choroideremia
Choroideremia is an X-linked chorioretinal dystrophy characterized by the diffuse, progressive degeneration of the RPE, photoreceptors, and choriocapillaris. A mutation in the CHM gene causes it. On fundus examination, the earliest manifestation is widespread pigment clumping at the level of the RPE, which is distinct from the characteristic perivascular bone-spicule pigment clumping seen in retinitis pigmentosa. Subsequently, patients develop well-defined regions of atrophy with visible underlying sclera and large choroidal vessels, most commonly in the postequatorial region just outside the vascular arcades. Typically, the fovea is not involved or affected very late. Some cases may clinically simulate the gyrate atrophy of the retina and retinal pigment epithelium.[68]
X-linked juvenile retinoschisis
X-linked juvenile retinoschisis is a rare congenital disease of the retina caused by mutations in the RS1 gene, which encodes retinoschisis. On fundoscopic examination, 98-100% of patients have foveal schisis, noted as a spoke wheel pattern radiating from the fovea. Subretinal linear fibrosis, pigmentation, white retinal flecks, vascular attenuation, and vascular sheathing may also be present. Peripheral retinoschisis is seen in around half of the patients. The intraretinal split is thought to be at the nerve fiber layer, as seen in the ophthalmoscope.[69]
Autosomal dominant optic atrophy
Autosomal dominant optic atrophy is commonly associated with mutations in the OPA1 gene located on chromosome 3q28-q29. Optic disc atrophy typically shows focal, wedged-shaped temporal optic atrophy. However, diffuse atrophy may be present.[70]
Evaluation
Fundus Photo and Ultrawide Field Fundus Imaging
Color fundus photos and ultrawide field fundus imaging are important for the documentation and analysis of the progression of the disease. Specifically, ultrawide field imaging may help document the retinal dystrophies affecting the peripheral fundus, including retinitis pigmentosa and gyrate atrophy of the retina and RPE.[71][72][73]
Optical Coherence Tomography
Spectral-domain optical coherence tomography (OCT) provides valuable information for diagnosing and monitoring the disease's progression. In retinitis pigmentosa, thinning of retinal layers, especially outer retinal layers, is commonly seen. The thinning progresses towards the macula, showing the foveal layers' sparing until the advanced stages.[74] OCT also detects thinning of the outer nuclear layer, loss or disruption of the ellipsoid zone and RPE, cystoid macular edema (CME), vitreomacular traction, macular hole, choroidal neovascular membrane, subretinal deposits, macular thinning, and epiretinal membrane.[75] OCT findings in various retinal dystrophies include:
- Retinitis pigmentosa: The subfoveal ellipsoid zone is usually preserved until late in the disease course; the loss of the ellipsoid zone and thinning of the outer nuclear layer are seen outside the fovea.
- Stargardt disease: Findings include thickened external limiting membrane (ELM) in early stages [76], thinning of outer nuclear layer, disruption of subfoveal ELM, ellipsoid zone, thinning of RPE and choriocapillaris, macular thinning, optical gap in the outer retina due to loss of ellipsoid zone in some phenotypes, hyper-reflective foci in the choroid, and subretinal deposit (pisciform fleck) which may sometimes migrate intraretinally (see Image. Optical Coherence Tomography of the Macula).[77][8]
- Rod monochromatism: Findings include subfoveal loss of ellipsoid zone, an optically empty cavity at the outer retina, loss of RPE, foveal hypoplasia, and loss of outer retinal layers with foveal thinning.[78]
- Best vitelliform macular dystrophy: Findings include a thickened middle highly reflective layer (HRL) [79], vitelliform deposit (homogenous hyperreflective material) subretinally and subretinal hyporeflective spaces in patients in pseudohypopyon stage and elongated outer photoreceptors over the area with subretinal hyporeflective space.[28]
A few-shot learning (FSL) model used the following criteria to define 3 types of retinal dystrophies:
- Cone or cone-rod lesions: Photoreceptor layer disruption and thinning of the sensory retina at the fovea
- Rod-cone lesions: Photoreceptor layer disruption and thinning of the sensory retina at the fovea outside the fovea with relative preservation of the structure at the fovea
- Extensive lesions [80]
Additionally, no changes in OCT may be noted in cases of CSNB with normal fundi. Fundus albipunctatus due to RDH5 may show deposition of hyperreflective material below the ELM at the area of the white dots, and old patients may show subfoveal preserved ELM with focal loss of ellipsoid zone and debris-like material deep to the ELM.[81] In fundus albipunctatus, hyperreflective deposits are noted over RPE, corresponding to retinal flecks observed clinically.[82] Degenerative changes, including outer retinal tubulations and inner retinal pseudocysts, may be noted in retinal dystrophies, causing atrophy at or around the macula, including Stargardt disease, cone dystrophy, and maternally inherited diabetes and deafness.[83]
Optical Coherence Tomography Angiography
Optical coherence tomography angiography (OCTA) is a noninvasive imaging technique that has significantly enhanced the evaluation of retinal dystrophies by providing detailed visualization of the retinal and choroidal vasculature. In conditions such as retinitis pigmentosa and choroideremia, OCTA can reveal characteristic vascular changes, including reduced vascular density and alterations in the foveal avascular zone.[84] This modality aids in the identification of neovascularization and ischemic areas, which are critical for understanding disease progression and potential complications. Moreover, OCTA allows for monitoring treatment responses in clinical trials and therapeutic interventions, especially in choroidal neovascular membranes (CNVMs), enhancing personalized patient management. Performing OCTA alongside traditional OCT imaging facilitates a comprehensive assessment of structural and vascular changes in retinal dystrophies.[85]
Fundus Autofluorescence
Fundus autofluorescence (FAF) is a valuable imaging modality in assessing retinal dystrophies, particularly in visualizing the health of the RPE. It provides insights into the metabolic activity of the RPE and the presence of lipofuscin, which accumulates in various retinal degenerative conditions. In retinitis pigmentosa, normal fundus autofluorescence at the fovea is usually preserved, bordered by a hyper-autofluorescent ring (Robson-Holder ring) outside which an abnormal mottled hypoautofluorescence is seen.[86] Similar rings may also be noted in other retinal dystrophies, including:
- Sector retinitis pigmentosa
- Bull's eye maculopathy
- Leber congenital amaurosis
- Cone dystrophy
- Cone-rod dystrophy
- Best disease
- X-linked retinoschisis [86][87]
The Best disease demonstrates a highly hyperautofluorescent lesion at the fovea in the vitelliform stage.[64] The vitelliform lesions in the posterior pole in autosomal recessive bestrophinopathy are also highly hyperautofluorescent.[88] Stargardt disease is characterized by hypoautofluorescence at the area of macular atrophy, and the flecks are usually hyperautofluorescent, which typically spare the area around the optic disc (compared to pentosan polysulfate maculopathy that causes autofluorescence changes around the optic disc also).[40] Choroideremia is characterized by hyper-autofluorescence in the areas with visible sclera, and the macula usually has a stellate area with normal choriocapillaris and RPE.[87] Fundus albipunctata shows a grainy image and reduced background autofluorescence, and the white dots may show hyperautofluorescence.[81]
Fundus Fluorescein Angiogram
Stargardt disease is associated with dark choroid in at least 80% of cases. The areas of RPE atrophy at the macula due to cone dystrophy, Stargardt disease, and other retinal dystrophies cause window defects. Peripheral window defects are seen in areas of RPE atrophy in retinitis pigmentosa, gyrate atrophy, and other diseases. The vitelliform lesion in Best disease and pigments in retinitis pigmentosa may show block fluorescence. Typically, the macular edema in retinal dystrophies is nonleaking (not causing a petaloid leak in fluorescein angiogram or angiographically silent CME), hence more appropriately called maculoschisis or foveoschisis. Such CME may respond well to oral or topical carbonic anhydrase inhibitors. The retinal dystrophies associated with nonleaking macular edema include:
- Retinitis pigmentosa
- Gyrate atrophy of retina and RPE [71]
- X-linked retinoschisis
- Goldmann Favre syndrome or enhanced S-cone syndrome (NR2E3-related retinopathies)
- Choroideremia
- MIDD
- Cohen syndrome (rod-cone dystrophy) [89]
However, in some cases of retinitis pigmentosa, there may be clinical evidence of active intermediate uveitis associated with leaking CME, which responds well to periocular or oral steroids.[90][73] Fundus fluorescein angiogram also helps detect CNVMs, which can complicate many retinal dystrophies.
Visual Field Analysis
Peripheral visual field loss is common in rod-dominated disorders.[91] Mild constriction of peripheral fields is seen initially, progressing gradually to tunnel vision over the years. The visual field changes are usually symmetric in both eyes. Regular field assessments are mandatory in progressive diseases such as retinitis pigmentosa, especially if the patient drives a motor vehicle. All patients with retinitis pigmentosa must restrict night driving and eventually stop driving as the disease progresses. Regular field assessments also enable patients to understand their visual limitations, which are not realized otherwise. Though the central vision of retinitis pigmentosa cases may be excellent (20/20), they can come under the definition of legal blindness due to the visual field criteria (severely narrowed visual field).[92]
In cone-dominated disorders, peripheral field constriction is rarely seen and remains stable over the years if present. Progression of field defects rules out the diagnosis, and one must consider other differentials like cone-rod dystrophy or retinitis pigmentosa with a cone-rod pattern. Visual field defects in cone-rod dystrophies begin as central scotoma and involve the periphery in later stages.[91][34][91]
Color Vision
Retinitis pigmentosa patients maintain good color discrimination until the advanced stages when cones are affected. Color vision abnormalities are reported once the visual acuity declines below 20/40.
Color vision abnormalities are more reported in cone-dominated disorders. Complete achromats are congenitally color blind; however, they may be able to identify different pseudo-isochromatic color plates as they train themselves to identify different colors as shades of gray. Hence, higher-order color tests may be required for diagnosis. Blue cone monochromats, as the name suggests, preserve Tritan color discrimination and can be tested on higher-order color tests like Berson plates.[93]
Electrophysiology
The full-field electroretinogram (ERG) is a sensitive test for diagnosis. In the early stages of retinitis pigmentosa, diminished scotopic rod and combined responses are seen. As the disease advances, photopic responses are affected, and eventually, ERG becomes extinguished.
ERG is the mainstay in the diagnosis of the cone or cone-rod dystrophies. Decreased photopic and 30 Hz flicker ERG with delays in implicit times are seen. Multifocal ERGs will be severely diminished. Later in the course of the disease, scotopic responses are also reduced. However, reduced scotopic responses in the early stages are a pointer towards diagnosing retinitis pigmentosa. The differentiating feature of achromatopsia and blue monochromatism is that a cone signal can be obtained using a blue light flash on a yellow background in the latter.[94] Cases with Leber congenital amaurosis are known to have profoundly abnormal or extinguished ERG. ERG is also essential in differentiating retinitis pigmentosa from disorders like cone-rod dystrophy. In cases with CRD, marked reduction or absence of cone ERG responses in the presence of less reduction in rod responses are seen.
Electro-oculogram (EOG) is typically abnormal in Best disease and other retinal dystrophies caused by mutations in the bestrophin-1 gene.[95]
Adaptive Optics
Adaptive optics (AO) allows for high-resolution photoreceptor layer and retinal microstructure visualization. This technique enhances the contrast and resolution of images, enabling the detection of early retinal changes that may not be visible with conventional imaging methods. In conditions like retinitis pigmentosa and Stargardt disease, AO can reveal abnormal photoreceptor morphology and quantify cell density, providing insights into disease progression and potential treatment outcomes.[96][97] Furthermore, AO can facilitate the evaluation of localized changes in retinal structure associated with genetic mutations, aiding in personalized management approaches. As a noninvasive method, it holds promise for longitudinal studies that track disease evolution over time.[98]
Treatment / Management
Currently, retinal dystrophies are not curable. However, supportive treatment can improve the quality of life. Major options to consider in all cases include refraction, cataract surgery when indicated, and referral for low vision aids. Patients with CME benefit from topical or systemic carbonic anhydrase inhibitors.[99] CME associated with intraocular inflammation may benefit from periocular or systemic steroids. The patients should avoid smoking and retinotoxic medications. Anti-VEGF (anti-vascular endothelial growth factor) agents are indicated in retinal dystrophies complicated by CNVM.
Patients with night vision problems benefit from night vision aids or commonly used devices like flashlights. Cases with cone dystrophies with photosensitivity benefit from tinted lenses, mainly orange or red lenses, since rod photoreceptors are less sensitive to orange and red lights.[100] Red-tinted soft contact lenses are an alternative to glasses. Low vision aids may help enhance visual acuity.
Gene Therapy
The retina is a good target for genetic manipulation due to various reasons: target cells (photoreceptors) are easily accessible for surgery and monitoring, the blood-retinal barrier provides an ocular immune privilege, and the cell population is static, which requires a small number of therapeutics to be administered. Gene therapy may work through various methods, including:
- Gene replacement
- Gene silencing
- Gene editing
- Therapeutic oligonucleotides [101]
Subretinal injections are a preferred mode of delivery for gene therapy as the agent reaches RPE and photoreceptors more precisely. Compared to intravitreal injections, the immunological response is well controlled. Several clinical trials are ongoing. In 2017, the USFDA approved subretinal injection of voretigene neparvovec for RPE65-associated Leber congenital amaurosis.[102] The gene therapy needs viral or nonviral carriers or vectors to deliver the drug to the target tissue. Viral vectors include adeno-associated virus and lentivirus.[103] Nonviral vectors include delivery systems based on nanomaterials, lipids, and magnetic nanoparticles.[101] Gene editing technologies include clustered, regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins.[103] Genetic targets for various retinal dystrophies include:(A1)
- Retinitis pigmentosa: RPGR, PDE6A, PDE6B, MERTK, RHO, RLBP1
- Achromatopsia: CNGA3, CNGB3
- Leber congenital amaurosis: CEP290, RPE65, GUCY2D
- Usher syndrome: MYO7A
- Stargardt disease: ABCA4
- Choroideremia: CHM
- Bietti crystalline dystrophy: CYP4V2
- X-linked retinoschisis: RS1 [101][103]
Stem Cell Therapy
Human pluripotent stem cells were first cultured in 1998 and were seen to have the potential to differentiate into endodermal, mesodermal, and ectodermal lineages. The retina is a good target for stem cell therapy for reasons similar to gene therapy. Moreover, the limited size of the retina requires smaller quantities of therapeutic tissue compared to other organs. Several ongoing trials mainly focus on diseases like Stargardt disease and retinitis pigmentosa.[104](A1)
Retinal Prostheses
Retinal prosthesis devices replace phototransduction within the eyes of individuals lacking photoreceptors for diseases, eg, retinitis pigmentosa. Normally, photoreceptors contain light-sensitive pigments that trigger phototransduction, which generates neuronal signals in the presence of light stimuli. The middle layers of the retina process these signals before they reach ganglion cells. These retinal ganglion cells form the optic nerve and transmit these signals to the visual cortex. In cases where the outer retinal layer/photoreceptors are lost, the retinal prosthesis transmits the signals to the inner layers.
In 2013, the FDA approved Argus II retinal prosthesis for late-stage retinitis pigmentosa. Many other retinal prostheses are under trial worldwide to explore their use in conditions like severe age-related macular degeneration, cone-rod dystrophy, and choroideremia. Argus II is composed of 60 electrodes that are implanted epiretinally. It has 3 external and 3 internal components. External components include a video camera mounted on a pair of glasses, a visual processing unit, and a coil attached to the sidearm of glasses. Internal components are an internal coil and internal processing unit, which is within a casing that is sutured to the sclera.
Retinal prosthesis is indicated in individuals with retinitis pigmentosa older than 25, with light perception or worse vision, and a previous history of useful form vision. Patients must also be highly motivated to comply with postoperative follow-up and rehabilitation. Complications reported include retinal detachment, choroidal effusion, hypotony, endophthalmitis, and implant dislocation.[105] As new therapies continue to be discovered, clinicians must recognize their clinical characteristics to determine whether patients may benefit from a specific treatment.
Genetic Consultation
Genetic counseling should be arranged once the diagnosis of retinitis pigmentosa or other retinal dystrophies is suspected. The consultation aims to confirm the diagnosis and mode of inheritance. The prognosis of the disease may also be known if the genetic mutation is known. Counseling involves taking a history and conducting a clinical examination in addition to clinical investigations and molecular genetic diagnosis. A detailed pedigree is essential for a complete workup. Once the mode of inheritance is known, tailored genetic testing must be done. Currently, at least 270 genes associated with retinal dystrophies can be tested.
Counseling must address the risk of progression and the current changes in lifestyle required. Patients must be aware of the risk of disease transmission in future generations. Pedigree charts help in assessing the mode of inheritance. For instance, in autosomal recessive families, in which both parents are carriers, each child has a 25% risk of inheritance. An individual with autosomal recessive disease has a small risk of transmitting the disease to his offspring, depending on the carrier state of the population. In such circumstances, consanguineous marriages must be avoided.
All patients must be offered a referral to support services. This is particularly important when dealing with issues like vocational rehabilitation training and schooling of a visually impaired child.
Mental Health Therapy
Psychological support and psychiatry consultation are essential parts of management as the diagnosis of retinal dystrophies may be overwhelming for patients, and such patients may have a higher risk of depression and anxiety.[106]
Nutritional Supplements
Supplementation of vitamin B6 and an arginine-restricted diet may help some patients with gyrate atrophy. Vitamin A has been used with variable success in many patients with retinitis pigmentosa. It has been noted that vitamin A supplementation should be avoided in patients with Stargardt disease as it may lead to increased production of lipofuscin, which is a metabolic byproduct of vitamin A.[107]
The following are treatable forms of retinitis pigmentosa and their proposed management:
- The Bassen-Kornzweig disease may be treated with vitamins A, E, and K supplementation and a low-fat diet [108]
- Friedreich-like ataxia associated with retinitis pigmentosa may benefit from vitamin E therapy [108][100]
- Refsum disease patients need a phytanic acid-restricted diet [109]
Other promising options in managing retinal dystrophies include intravitreal injections of ciliary neurotrophic factor and valproic acid.[110][111] Syndromic retinal dystrophies require interprofessional collaboration for holistic patient care. With optimum management, the quality of life can be improved in most patients with retinal dystrophies.(A1)
Differential Diagnosis
Each retinal dystrophy phenotype is often confused with other genetic or acquired disorders. Misdiagnosis is common and must be avoided, as it has a great bearing on genetic and prognostic counseling. Acquired causes can be treated in many cases; therefore, timely diagnosis is necessary.
Retinitis pigmentosa must be differentiated from other conditions causing retinal pigmentation, eg, rubella retinopathy, syphilis, autoimmune paraneoplastic retinopathy, and drug toxicities. Quinine overdose can cause sudden loss of vision and manifest with optic disc pallor and vessel attenuation. It can be misdiagnosed as the sine pigmento stage of retinitis pigmentosa. ERG in quinine overdose has a negative configuration, affecting the b wave more than a wave.[112]
ABCA4 gene has extensive phenotypic heterogeneity, and mutations can cause various diseases, including:
- Autosomal recessive Stargardt disease
- Fundus flavimaculatus (autosomal recessive, AR)
- Cone rod dystrophy 3 (AR)
- Retinitis pigmentosa 19 (AR)
- Early onset severe retinal dystrophy (AR)
- Age-related macular degeneration 2 (autosomal dominant, AD)
Pigmented paravenous retinochoroidal atrophy or pigmented paravenous chorioretinal atrophy also manifests with retinal pigmentation along the retinal veins. Retinal pigmentation is mainly an incidental finding associated with tuberculosis, syphilis, rubeola, and meningoencephalitis. ERG responses in these conditions are only mild to moderate abnormal.
Occasionally, traumatic retinopathy and DUSN can be misdiagnosed as unilateral retinitis pigmentosa. Retinitis pigmentosa may be asymmetric initially; hence, long-term follow-up is needed before diagnosing unilateral retinitis pigmentosa. In all cases of so-called unilateral retinitis pigmentosa or unilateral pigmentary retinopathy (UPR), other causes of pigmentary retinopathy must be excluded. Other causes of pigmentary retinopathy (pseudo-retinitis pigmentosa) include:
- Drugs, including phenothiazines (thioridazine), chloroquine, hydroxychloroquine
- Trauma, including traumatic retinopathy, siderosis, and retinal detachment
- Infections including toxoplasmosis, rubella, syphilis, tuberculosis, retinitis including cytomegaloviral retinitis and acute retinal necrosis, measles
- Inflammations, including acute zonal occult outer retinopathy, DUSN, healed posterior uveitis, and severe retinal vasculitis,
- Retinal detachment may spontaneously resolve (self-settled retinal detachment), giving an appearance of pigmentary retinopathy [113]
- Autoimmune retinopathy (AIR) can be nonparaneoplastic (npAIR), including CAR, MAR, PON, and BDUMP, or paraneoplastic (pAIR)
Advanced cases of choroideremia, Stargardt macular dystrophy, and cone-rod dystrophy are usually misdiagnosed as retinitis pigmentosa. Retinitis punctata albescens and fundus albipunctatus have overlapping fundus images. Patients with fundus albipunctatus are usually asymptomatic, and fundus findings are incidental. Rarely, they may complain of night blindness early in childhood without progression. However, retinitis punctata albescens is a progressive disease with worse symptoms and a reduction in the field of vision over time. However, fundus albipunctatus and retinitis punctata albescens might denote a spectrum of diseases caused by the same or similar mutations of genes, including RLBP1.[114] Mutations in RLBP1 may result in multiple phenotypes, including:
- Fundus albipunctatus (AD, AR)
- Retinitis punctata albescens(AD, AR)
- Newfoundland rod-cone dystrophy
- Bothnia retinal dystrophy (AR)
Electrophysiological characteristics of most diseases can help narrow the diagnosis in phenotypically similar conditions.
Prognosis
Retinitis pigmentosa, Leber congenital amaurosis, and cone-rod dystrophies are generally progressive. Early age of onset is usually associated with poor prognosis in retinitis pigmentosa and LCA. However, conditions like congenital stationary night blindness and achromatism are nonprogressive.[115] Proper counseling regarding the disease and its progression can help the patient and family cope with the challenges. Retinal pattern dystrophies and the 'Best disease' usually have good visual prognoses.[28] The life expectancy of people with retinal dystrophy is generally normal except for syndromic cases or cases with systemic disease.
Complications
Retinitis pigmentosa progresses at a varied rate, depending on various parameters, including the gene involved, family history, and age of onset, from peripheral constriction of fields to tunnel vision and gradual vision loss. Early onset of visually significant cataracts is common. Risk factors associated with cataract surgery are more prevalent in cases with retinal dystrophy than in the general population, including zonular weakness, capsular phimosis, early onset of posterior capsule opacification, and cystoid macular edema.[116] The intraretinal hyporefective spaces on macular OCT in retinitis pigmentosa, gyrate atrophy, X-linked retinoschisis, and Goldmann-Favre disease may not cause a petaloid leak on fundus fluorescein angiography and is better termed as "foveoschisis" or maculoschisis.[71][63][71] Vertical tissue bridges are seen on OCT. These cases may respond to topical, including dorzolamide or oral acetazolamide carbonic anhydrase inhibitors. Defects in color and peripheral vision may cause challenges during driving or moving. People with vision loss from retinal dystrophies may experience depression, which can include feelings of sadness, loss of interest, fatigue, weight loss or gain, and low energy.[117]
Deterrence and Patient Education
The diagnosis of retinal dystrophy causes a lot of anxiety to the patient and their family. Patients who are informed about the diagnosis are often apprehensive about losing their vision, adding to psychological stress. Except for LCA, most conditions are usually slowly progressive, and some have a stationary course. Another misconception is that retinal dystrophies are not treatable. They are incurable. However, supportive treatments, such as low vision assessment, support services, and visual rehabilitation, can significantly improve a patient's lifestyle.
Enhancing Healthcare Team Outcomes
An interprofessional team approach is critical for enhancing patient-centered care, outcomes, and safety in managing retinal dystrophies. Ophthalmologists lead the clinical diagnosis, utilizing advanced imaging and diagnostic tools, while geneticists and genetic nurse counselors play pivotal roles in ordering and interpreting genetic tests. They provide vital counseling to patients and their families regarding the diagnosis, prognosis, and genetic inheritance. Advanced practitioners, nurses, and pharmacists contribute by managing patient education, low vision aids, and coordinating rehabilitation services. Effective communication among these professionals ensures seamless care coordination, improves team performance, and enhances patient outcomes by addressing both medical and psychosocial needs comprehensively.
Media
(Click Image to Enlarge)
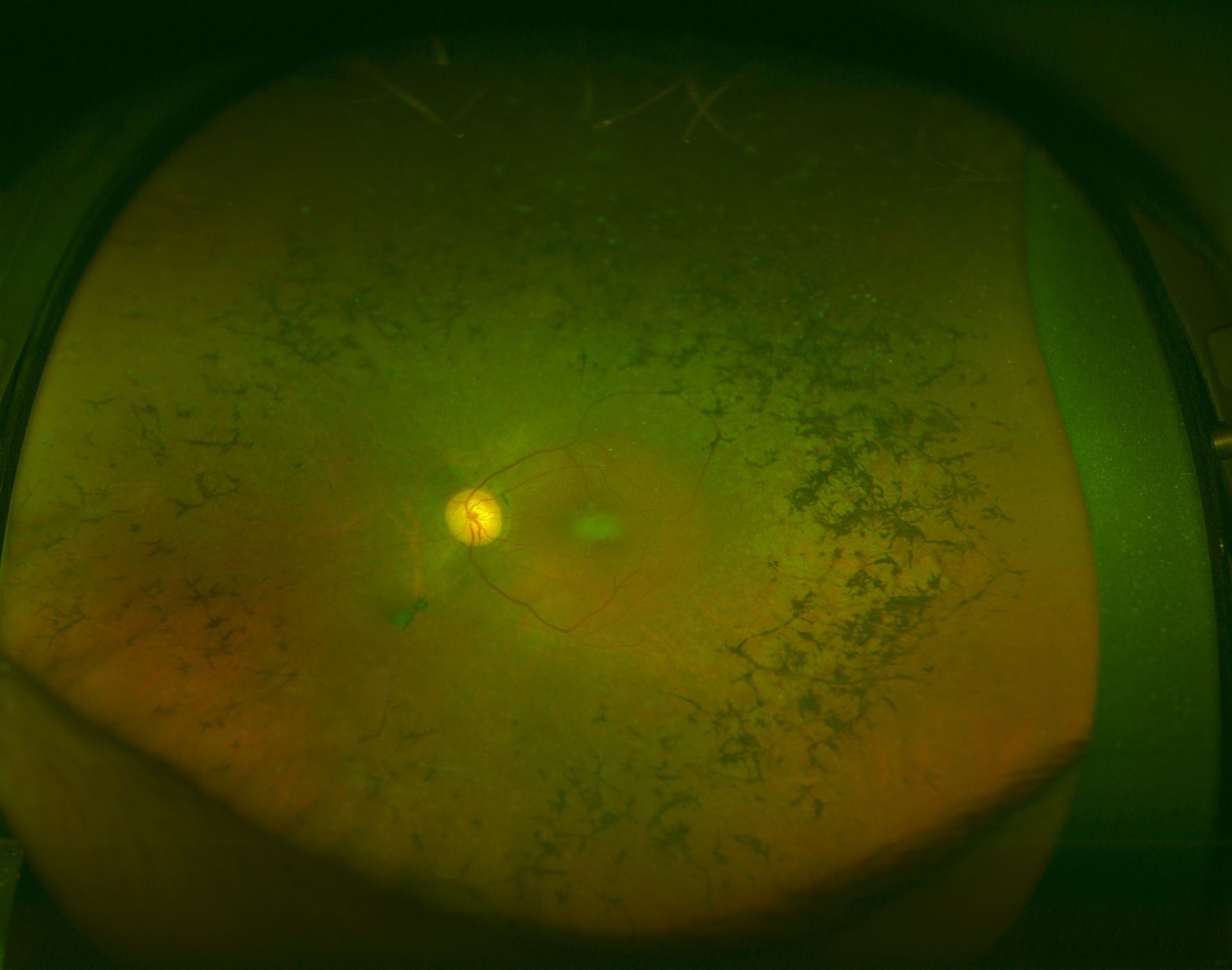
Retinitis Pigmentosa: The fundus appearance of RP classically includes a triad of retinal vessel attenuation, waxy pallor of the optic disc, and bone spicule intraretinal pigmentation.
Courtesy of K Tripathy, MD
(Click Image to Enlarge)
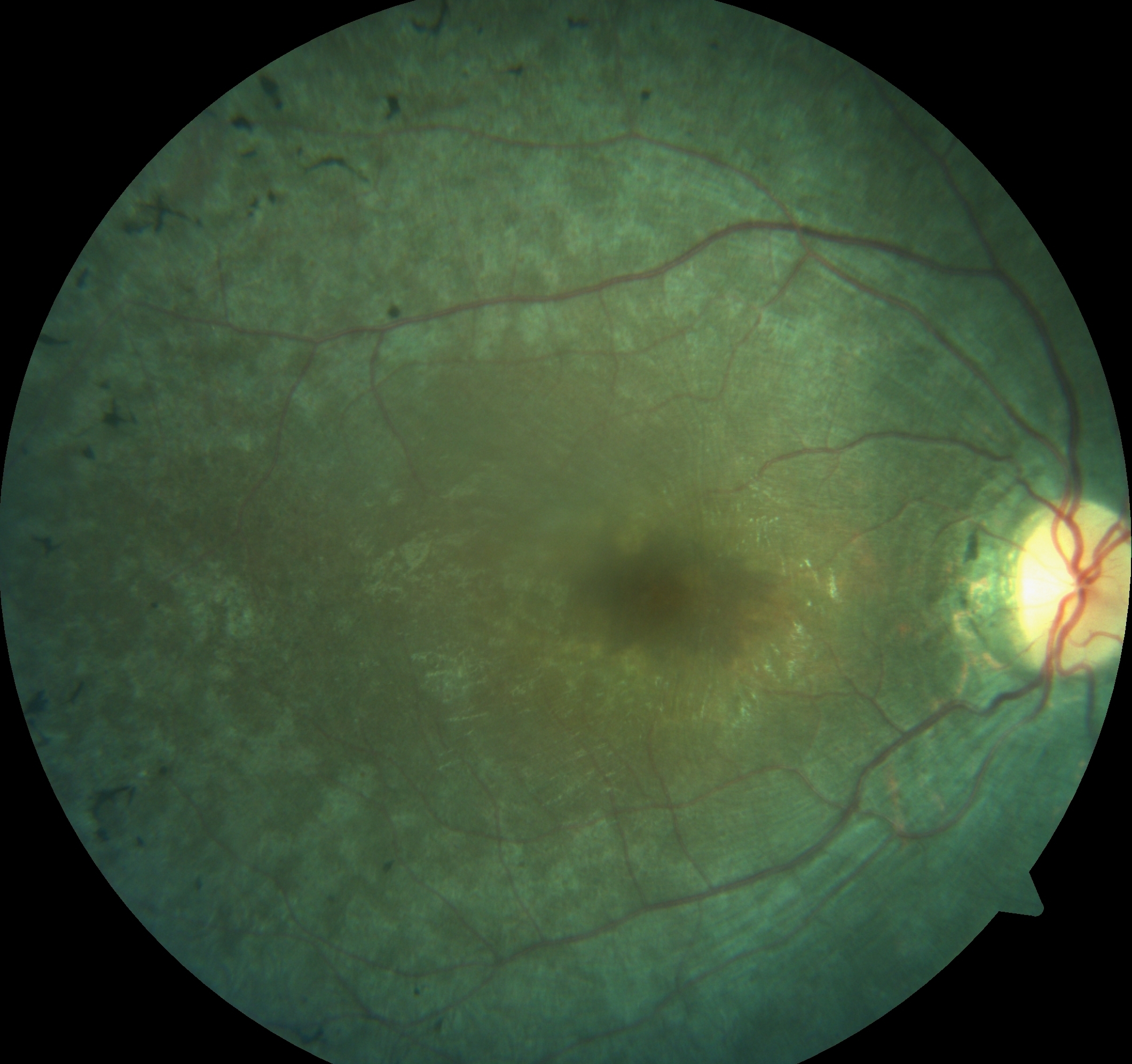
Pigmentary Retinopathy. Pigmentary retinopathy in a patient with Bardet Biedl syndrome.
Tripathy K, Chawla R, Sarkar S. Girl with polydactyly and pigmentary retinopathy. J Paediatr Child Health. 2017;53(4):424. doi:10.1111/jpc.1_13323.
(Click Image to Enlarge)

Stargardt Disease. Flecks and macular atrophy in a patient with autosomal recessive Stargardt disease.
Contributed by K Tripathy, MD
(Click Image to Enlarge)
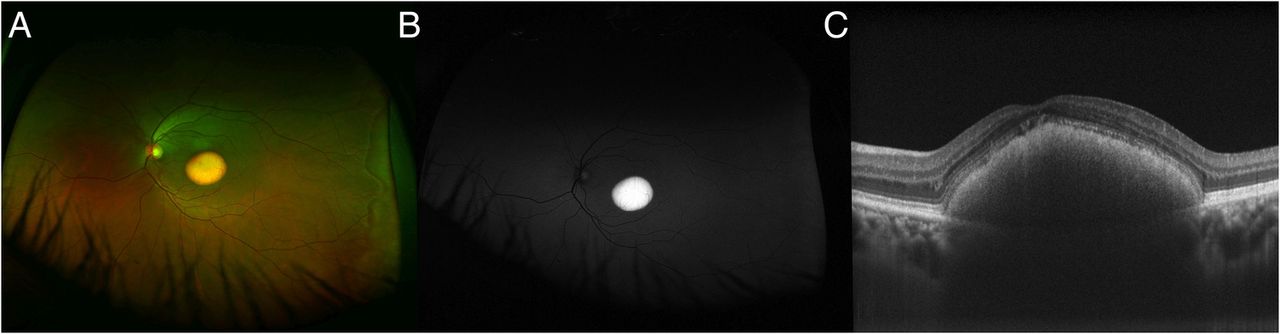
Imaging in Best Disease. Image A. Pseudocolor ultrawide field image, Image B. Fundus autofluorescence image, Image C. Optical coherence tomography of the macula.
Tripathy K, Chawla R, Mittal K, Temkar S. Egg yolk in the eye: an ultrawide field evaluation. BMJ Case Rep. 2016;2016:bcr2016214358. doi: 10.1136/bcr-2016-214358.
(Click Image to Enlarge)
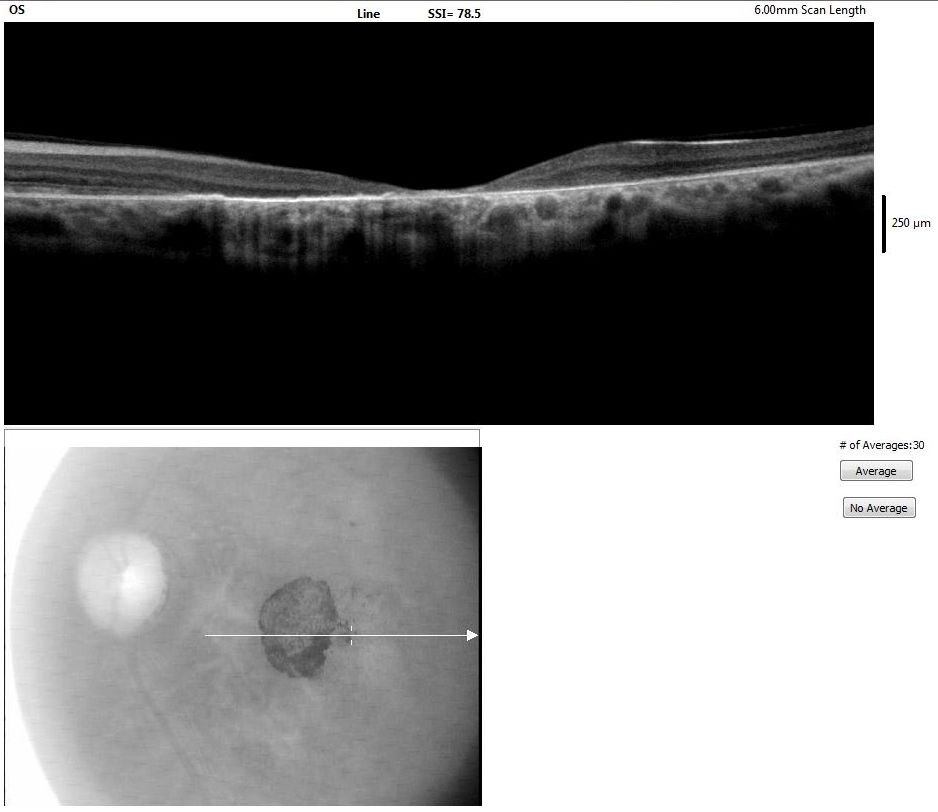
Optical Coherence Tomography of the Macula. Tomography image of the macula in a patient with advanced Stargardt disease showing foveal thinning.
Contributed by K Tripathy, MD
References
Corradetti G, Verma A, Tojjar J, Almidani L, Oncel D, Emamverdi M, Bradley A, Lindenberg S, Nittala MG, Sadda SR. Retinal Imaging Findings in Inherited Retinal Diseases. Journal of clinical medicine. 2024 Apr 3:13(7):. doi: 10.3390/jcm13072079. Epub 2024 Apr 3 [PubMed PMID: 38610844]
García Bohórquez B, Aller E, Rodríguez Muñoz A, Jaijo T, García García G, Millán JM. Updating the Genetic Landscape of Inherited Retinal Dystrophies. Frontiers in cell and developmental biology. 2021:9():645600. doi: 10.3389/fcell.2021.645600. Epub 2021 Jul 13 [PubMed PMID: 34327195]
Huang XF, Huang F, Wu KC, Wu J, Chen J, Pang CP, Lu F, Qu J, Jin ZB. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015 Apr:17(4):271-8. doi: 10.1038/gim.2014.138. Epub 2014 Nov 6 [PubMed PMID: 25356976]
Daich Varela M, Cabral de Guimaraes TA, Georgiou M, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: current management and clinical trials. The British journal of ophthalmology. 2022 Apr:106(4):445-451. doi: 10.1136/bjophthalmol-2020-318483. Epub 2021 Mar 12 [PubMed PMID: 33712480]
Kolb H, Fernandez E, Jones B, Nelson R, Fu Y. Phototransduction in Rods and Cones. Webvision: The Organization of the Retina and Visual System. 1995:(): [PubMed PMID: 21413414]
Tatour Y, Ben-Yosef T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics (Basel, Switzerland). 2020 Oct 2:10(10):. doi: 10.3390/diagnostics10100779. Epub 2020 Oct 2 [PubMed PMID: 33023209]
Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, Braun TA, Mullins RF, Scheetz TE, Sheffield VC, Tucker BA. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology. 2017 Sep:124(9):1314-1331. doi: 10.1016/j.ophtha.2017.04.008. Epub 2017 May 27 [PubMed PMID: 28559085]
Kohli P, Tripathy K, Kaur K. Stargardt Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 36508525]
Elnahry AG, Tripathy K. Gyrate Atrophy of the Choroid and Retina. StatPearls. 2024 Jan:(): [PubMed PMID: 32491691]
Retinitis Pigmentosa, O'Neal TB,Luther EE,,, 2018 Jan [PubMed PMID: 30137803]
Tripathy K, Chawla R, Sarkar S. Girl with polydactyly and pigmentary retinopathy. Journal of paediatrics and child health. 2017 Apr:53(4):424. doi: 10.1111/jpc.1_13323. Epub [PubMed PMID: 28370859]
Ahmed H, Sierpina DI, Khazaeni L. Retinal Pattern Dystrophy. StatPearls. 2024 Jan:(): [PubMed PMID: 35881734]
Bunker CH, Berson EL, Bromley WC, Hayes RP, Roderick TH. Prevalence of retinitis pigmentosa in Maine. American journal of ophthalmology. 1984 Mar:97(3):357-65 [PubMed PMID: 6702974]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Kohl S, Jägle H, Wissinger B, Zobor D. Achromatopsia. GeneReviews(®). 1993:(): [PubMed PMID: 20301591]
Lin S, Vermeirsch S, Pontikos N, Martin-Gutierrez MP, Daich Varela M, Malka S, Schiff E, Knight H, Wright G, Jurkute N, Simcoe MJ, Yu-Wai-Man P, Moosajee M, Michaelides M, Mahroo OA, Webster AR, Arno G. Spectrum of Genetic Variants in the Most Common Genes Causing Inherited Retinal Disease in a Large Molecularly Characterized United Kingdom Cohort. Ophthalmology. Retina. 2024 Jul:8(7):699-709. doi: 10.1016/j.oret.2024.01.012. Epub 2024 Jan 12 [PubMed PMID: 38219857]
Pontikos N, Arno G, Jurkute N, Schiff E, Ba-Abbad R, Malka S, Gimenez A, Georgiou M, Wright G, Armengol M, Knight H, Katz M, Moosajee M, Yu-Wai-Man P, Moore AT, Michaelides M, Webster AR, Mahroo OA. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology. 2020 Oct:127(10):1384-1394. doi: 10.1016/j.ophtha.2020.04.008. Epub 2020 Apr 16 [PubMed PMID: 32423767]
Small KW, Puech B, Mullen L, Yelchits S. North Carolina macular dystrophy phenotype in France maps to the MCDR1 locus. Molecular vision. 1997 Jan 2:3():1 [PubMed PMID: 9238090]
Small KW. North Carolina macular dystrophy, revisited. Ophthalmology. 1989 Dec:96(12):1747-54 [PubMed PMID: 2622620]
Small KW, DeLuca AP, Whitmore SS, Rosenberg T, Silva-Garcia R, Udar N, Puech B, Garcia CA, Rice TA, Fishman GA, Héon E, Folk JC, Streb LM, Haas CM, Wiley LA, Scheetz TE, Fingert JH, Mullins RF, Tucker BA, Stone EM. North Carolina Macular Dystrophy Is Caused by Dysregulation of the Retinal Transcription Factor PRDM13. Ophthalmology. 2016 Jan:123(1):9-18. doi: 10.1016/j.ophtha.2015.10.006. Epub 2015 Oct 24 [PubMed PMID: 26507665]
Hussels IE, Morton NE. Pingelap and Mokil Atolls: achromatopsia. American journal of human genetics. 1972 May:24(3):304-9 [PubMed PMID: 4555088]
Manley A, Meshkat BI, Jablonski MM, Hollingsworth TJ. Cellular and Molecular Mechanisms of Pathogenesis Underlying Inherited Retinal Dystrophies. Biomolecules. 2023 Feb 1:13(2):. doi: 10.3390/biom13020271. Epub 2023 Feb 1 [PubMed PMID: 36830640]
Ben-Yosef T. Inherited Retinal Diseases. International journal of molecular sciences. 2022 Nov 3:23(21):. doi: 10.3390/ijms232113467. Epub 2022 Nov 3 [PubMed PMID: 36362249]
Hollingsworth TJ, Gross AK. Defective trafficking of rhodopsin and its role in retinal degenerations. International review of cell and molecular biology. 2012:293():1-44. doi: 10.1016/B978-0-12-394304-0.00006-3. Epub [PubMed PMID: 22251557]
Lewis TR, Makia MS, Castillo CM, Al-Ubaidi MR, Naash MI, Arshavsky VY. Photoreceptor Disc Enclosure Is Tightly Controlled by Peripherin-2 Oligomerization. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2021 Apr 21:41(16):3588-3596. doi: 10.1523/JNEUROSCI.0041-21.2021. Epub 2021 Mar 11 [PubMed PMID: 33707293]
Chen HY, Welby E, Li T, Swaroop A. Retinal disease in ciliopathies: Recent advances with a focus on stem cell-based therapies. Translational science of rare diseases. 2019 Jul 4:4(1-2):97-115. doi: 10.3233/TRD-190038. Epub 2019 Jul 4 [PubMed PMID: 31763178]
Level 3 (low-level) evidenceBujakowska KM, Liu Q, Pierce EA. Photoreceptor Cilia and Retinal Ciliopathies. Cold Spring Harbor perspectives in biology. 2017 Oct 3:9(10):. doi: 10.1101/cshperspect.a028274. Epub 2017 Oct 3 [PubMed PMID: 28289063]
Level 3 (low-level) evidenceYang S, Zhou J, Li D. Functions and Diseases of the Retinal Pigment Epithelium. Frontiers in pharmacology. 2021:12():727870. doi: 10.3389/fphar.2021.727870. Epub 2021 Jul 28 [PubMed PMID: 34393803]
Tripathy K,Salini B, Best Disease StatPearls. 2021 Jan [PubMed PMID: 30725975]
Gartner S, Henkind P. Pathology of retinitis pigmentosa. Ophthalmology. 1982 Dec:89(12):1425-32 [PubMed PMID: 7162785]
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Progress in retinal and eye research. 2018 Jan:62():24-37. doi: 10.1016/j.preteyeres.2017.08.004. Epub 2017 Sep 27 [PubMed PMID: 28962928]
Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Progress in retinal and eye research. 1998 Apr:17(2):175-205 [PubMed PMID: 9695792]
Kolb H, Fernandez E, Jones B, Nelson R, Jones BW, Marc RE, Pfeiffer RL. Retinal Degeneration, Remodeling and Plasticity. Webvision: The Organization of the Retina and Visual System. 1995:(): [PubMed PMID: 29493934]
Michaelides M, Hardcastle AJ, Hunt DM, Moore AT. Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Survey of ophthalmology. 2006 May-Jun:51(3):232-58 [PubMed PMID: 16644365]
Level 3 (low-level) evidenceHamel CP, Cone rod dystrophies. Orphanet journal of rare diseases. 2007 Feb 1; [PubMed PMID: 17270046]
Bonilha VL, Rayborn ME, Bell BA, Marino MJ, Fishman GA, Hollyfield JG. Retinal Histopathology in Eyes from a Patient with Stargardt disease caused by Compound Heterozygous ABCA4 Mutations. Ophthalmic genetics. 2016 Jun:37(2):150-60. doi: 10.3109/13816810.2014.958861. Epub 2014 Sep 29 [PubMed PMID: 25265374]
Birnbach CD, Järveläinen M, Possin DE, Milam AH. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994 Jul:101(7):1211-9 [PubMed PMID: 8035984]
Level 3 (low-level) evidenceO'Gorman S,Flaherty WA,Fishman GA,Berson EL, Histopathologic findings in Best's vitelliform macular dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960). 1988 Sep; [PubMed PMID: 3415551]
Level 3 (low-level) evidenceWeingeist TA, Kobrin JL, Watzke RC. Histopathology of Best's macular dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960). 1982 Jul:100(7):1108-14 [PubMed PMID: 7092654]
Level 3 (low-level) evidenceMichaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. The British journal of ophthalmology. 2004 Feb:88(2):291-7 [PubMed PMID: 14736794]
Margines JB, Hobbs SD. Pentosan Polysulfate Maculopathy. StatPearls. 2024 Jan:(): [PubMed PMID: 36944010]
DIDANOSINE RETINAL TOXICITY., Haug SJ,Wong RW,Day S,Choudhry N,Sneed S,Prasad P,Read S,McDonald RH,Agarwal A,Davis J,Sarraf D,, Retina (Philadelphia, Pa.), 2016 Dec [PubMed PMID: 28005674]
Singh D, Tripathy K. Cancer-Associated Retinopathy. StatPearls. 2024 Jan:(): [PubMed PMID: 35201711]
Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F. Retinitis pigmentosa: genes and disease mechanisms. Current genomics. 2011 Jun:12(4):238-49. doi: 10.2174/138920211795860107. Epub [PubMed PMID: 22131869]
Grover S, Fishman GA, Anderson RJ, Tozatti MS, Heckenlively JR, Weleber RG, Edwards AO, Brown J Jr. Visual acuity impairment in patients with retinitis pigmentosa at age 45 years or older. Ophthalmology. 1999 Sep:106(9):1780-5 [PubMed PMID: 10485550]
Level 2 (mid-level) evidenceVerbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Progress in retinal and eye research. 2018 Sep:66():157-186. doi: 10.1016/j.preteyeres.2018.03.005. Epub 2018 Mar 27 [PubMed PMID: 29597005]
Lee EK, Lee SY, Ma DJ, Yoon CK, Park UC, Yu HG. RETINITIS PIGMENTOSA SINE PIGMENTO: Clinical Spectrum and Pigment Development. Retina (Philadelphia, Pa.). 2022 Apr 1:42(4):807-815. doi: 10.1097/IAE.0000000000003367. Epub [PubMed PMID: 34907125]
Mukhopadhyay R, Holder GE, Moore AT, Webster AR. Unilateral retinitis pigmentosa occurring in an individual with a germline mutation in the RP1 gene. Archives of ophthalmology (Chicago, Ill. : 1960). 2011 Jul:129(7):954-6. doi: 10.1001/archophthalmol.2011.171. Epub [PubMed PMID: 21746989]
Level 3 (low-level) evidenceAgarwal R, Tripathy K, Bandyopadhyay G, Basu K. Mizuo-Nakamura phenomenon in an Indian male. Clinical case reports. 2019 Feb:7(2):401-403. doi: 10.1002/ccr3.1990. Epub 2019 Jan 13 [PubMed PMID: 30847219]
Level 3 (low-level) evidenceGodara P, Cooper RF, Sergouniotis PI, Diederichs MA, Streb MR, Genead MA, McAnany JJ, Webster AR, Moore AT, Dubis AM, Neitz M, Dubra A, Stone EM, Fishman GA, Han DP, Michaelides M, Carroll J. Assessing retinal structure in complete congenital stationary night blindness and Oguchi disease. American journal of ophthalmology. 2012 Dec:154(6):987-1001.e1. doi: 10.1016/j.ajo.2012.06.003. Epub 2012 Sep 7 [PubMed PMID: 22959359]
Gonzalez-Fernandez F, Kurz D, Bao Y, Newman S, Conway BP, Young JE, Han DP, Khani SC. 11-cis retinol dehydrogenase mutations as a major cause of the congenital night-blindness disorder known as fundus albipunctatus. Molecular vision. 1999 Dec 30:5():41 [PubMed PMID: 10617778]
Kellner U, Wissinger B, Tippmann S, Kohl S, Kraus H, Foerster MH. Blue cone monochromatism: clinical findings in patients with mutations in the red/green opsin gene cluster. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2004 Sep:242(9):729-35 [PubMed PMID: 15069569]
Level 2 (mid-level) evidenceGreenberg JP, Sherman J, Zweifel SA, Chen RW, Duncker T, Kohl S, Baumann B, Wissinger B, Yannuzzi LA, Tsang SH. Spectral-domain optical coherence tomography staging and autofluorescence imaging in achromatopsia. JAMA ophthalmology. 2014 Apr 1:132(4):437-45. doi: 10.1001/jamaophthalmol.2013.7987. Epub [PubMed PMID: 24504161]
Michaelides M, Johnson S, Simunovic MP, Bradshaw K, Holder G, Mollon JD, Moore AT, Hunt DM. Blue cone monochromatism: a phenotype and genotype assessment with evidence of progressive loss of cone function in older individuals. Eye (London, England). 2005 Jan:19(1):2-10 [PubMed PMID: 15094734]
Berson EL, Sandberg MA, Rosner B, Sullivan PL. Color plates to help identify patients with blue cone monochromatism. American journal of ophthalmology. 1983 Jun:95(6):741-7 [PubMed PMID: 6602551]
Tsang SH, Sharma T. Rod Monochromatism (Achromatopsia). Advances in experimental medicine and biology. 2018:1085():119-123. doi: 10.1007/978-3-319-95046-4_24. Epub [PubMed PMID: 30578497]
Level 3 (low-level) evidenceGardner JC, Michaelides M, Holder GE, Kanuga N, Webb TR, Mollon JD, Moore AT, Hardcastle AJ. Blue cone monochromacy: causative mutations and associated phenotypes. Molecular vision. 2009:15():876-84 [PubMed PMID: 19421413]
Iarossi G, Coppè AM, Passarelli C, Maltese PE, Sinibaldi L, Cappelli A, Cetola S, Novelli A, Buzzonetti L. Blue Cone Monochromatism with Foveal Hypoplasia Caused by the Concomitant Effect of Variants in OPN1LW/OPN1MW and GPR143 Genes. International journal of molecular sciences. 2021 Aug 10:22(16):. doi: 10.3390/ijms22168617. Epub 2021 Aug 10 [PubMed PMID: 34445325]
Cideciyan AV, Hufnagel RB, Carroll J, Sumaroka A, Luo X, Schwartz SB, Dubra A, Land M, Michaelides M, Gardner JC, Hardcastle AJ, Moore AT, Sisk RA, Ahmed ZM, Kohl S, Wissinger B, Jacobson SG. Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Human gene therapy. 2013 Dec:24(12):993-1006. doi: 10.1089/hum.2013.153. Epub 2013 Oct 30 [PubMed PMID: 24067079]
Flanders M, Lapointe ML, Brownstein S, Little JM. Keratoconus and Leber's congenital amaurosis: a clinicopathological correlation. Canadian journal of ophthalmology. Journal canadien d'ophtalmologie. 1984 Dec:19(7):310-4 [PubMed PMID: 6525578]
Level 3 (low-level) evidenceElder MJ. Leber congenital amaurosis and its association with keratoconus and keratoglobus. Journal of pediatric ophthalmology and strabismus. 1994 Jan-Feb:31(1):38-40 [PubMed PMID: 8195961]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Chao DL, Burr A, Pennesi M. RPE65-Related Leber Congenital Amaurosis / Early-Onset Severe Retinal Dystrophy. GeneReviews(®). 1993:(): [PubMed PMID: 31725251]
Toms M, Pagarkar W, Moosajee M. Usher syndrome: clinical features, molecular genetics and advancing therapeutics. Therapeutic advances in ophthalmology. 2020 Jan-Dec:12():2515841420952194. doi: 10.1177/2515841420952194. Epub 2020 Sep 17 [PubMed PMID: 32995707]
Level 3 (low-level) evidenceRuia S,Tripathy K, Fluorescein Angiography StatPearls. 2022 Jan; [PubMed PMID: 35015403]
Tripathy K,Chawla R,Mittal K,Temkar S, Egg yolk in the eye: an ultrawide field evaluation. BMJ case reports. 2016 Jan 27 [PubMed PMID: 26818692]
Level 3 (low-level) evidenceNipp GE, Lee T, Sarici K, Malek G, Hadziahmetovic M. Adult-onset foveomacular vitelliform dystrophy: epidemiology, pathophysiology, imaging, and prognosis. Frontiers in ophthalmology. 2023:3():1237788. doi: 10.3389/fopht.2023.1237788. Epub 2023 Aug 10 [PubMed PMID: 38983024]
Francis PJ, Schultz DW, Gregory AM, Schain MB, Barra R, Majewski J, Ott J, Acott T, Weleber RG, Klein ML. Genetic and phenotypic heterogeneity in pattern dystrophy. The British journal of ophthalmology. 2005 Sep:89(9):1115-9 [PubMed PMID: 16113362]
Tsang SH, Sharma T. Pattern Dystrophy. Advances in experimental medicine and biology. 2018:1085():91-96. doi: 10.1007/978-3-319-95046-4_17. Epub [PubMed PMID: 30578490]
Level 3 (low-level) evidenceAdam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, MacDonald IM, Hume S, Zhai Y, Xu M. Choroideremia. GeneReviews(®). 1993:(): [PubMed PMID: 20301511]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Sieving PA, MacDonald IM, Hoang S. X-Linked Congenital Retinoschisis. GeneReviews(®). 1993:(): [PubMed PMID: 20301401]
Ham M, Han J, Osann K, Smith M, Kimonis V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion. 2019 May:46():262-269. doi: 10.1016/j.mito.2018.07.006. Epub 2018 Aug 27 [PubMed PMID: 30165240]
Level 1 (high-level) evidenceTripathy K, Chawla R, Sharma YR, Gogia V. Ultrawide field fluorescein angiogram in a family with gyrate atrophy and foveoschisis. Oman journal of ophthalmology. 2016 May-Aug:9(2):104-6. doi: 10.4103/0974-620X.184529. Epub [PubMed PMID: 27433038]
Jena S, Tripathy K, Chawla R, Mansour AM. Ultrawide field imaging to document the progression of gyrate atrophy of the choroid and retina over 5 years. BMJ case reports. 2021 Aug 17:14(8):. doi: 10.1136/bcr-2021-244695. Epub 2021 Aug 17 [PubMed PMID: 34404670]
Level 3 (low-level) evidenceTripathy K, Chawla R, Venkatesh P, Vohra R, Sharma YR, Gogia V, Jain S, Behera A. Ultra-wide Field Fluorescein Angiography in Retinitis Pigmentosa with Intermediate Uveitis. Journal of ophthalmic & vision research. 2016 Apr-Jun:11(2):237-9. doi: 10.4103/2008-322X.183929. Epub [PubMed PMID: 27413510]
Hood DC, Ramachandran R, Holopigian K, Lazow M, Birch DG, Greenstein VC. Method for deriving visual field boundaries from OCT scans of patients with retinitis pigmentosa. Biomedical optics express. 2011 Apr 5:2(5):1106-14. doi: 10.1364/BOE.2.001106. Epub 2011 Apr 5 [PubMed PMID: 21559123]
Tripathy K, Sharma YR, Chawla R, Jain S, Behera A. Ultra-wide Field Imaging of an Operated Macular Hole in Gyrate Atrophy. Journal of ophthalmic & vision research. 2016 Jul-Sep:11(3):336-7. doi: 10.4103/2008-322X.188404. Epub [PubMed PMID: 27621797]
Burke TR, Yzer S, Zernant J, Smith RT, Tsang SH, Allikmets R. Abnormality in the external limiting membrane in early Stargardt disease. Ophthalmic genetics. 2013 Mar-Jun:34(1-2):75-7. doi: 10.3109/13816810.2012.707271. Epub 2012 Aug 7 [PubMed PMID: 22871184]
Level 3 (low-level) evidenceGómez-Benlloch A, Garrell-Salat X, Cobos E, López E, Esteve-Garcia A, Ruiz S, Vázquez M, Sararols L, Biarnés M. Optical Coherence Tomography in Inherited Macular Dystrophies: A Review. Diagnostics (Basel, Switzerland). 2024 Apr 24:14(9):. doi: 10.3390/diagnostics14090878. Epub 2024 Apr 24 [PubMed PMID: 38732293]
Thiadens AA, Somervuo V, van den Born LI, Roosing S, van Schooneveld MJ, Kuijpers RW, van Moll-Ramirez N, Cremers FP, Hoyng CB, Klaver CC. Progressive loss of cones in achromatopsia: an imaging study using spectral-domain optical coherence tomography. Investigative ophthalmology & visual science. 2010 Nov:51(11):5952-7. doi: 10.1167/iovs.10-5680. Epub 2010 Jun 23 [PubMed PMID: 20574029]
Ferrara DC,Costa RA,Tsang S,Calucci D,Jorge R,Freund KB, Multimodal fundus imaging in Best vitelliform macular dystrophy. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2010 Oct; [PubMed PMID: 20414784]
Zhao Q, Mai SW, Li Q, Huang GC, Gao MC, Yang WL, Wang G, Ma Y, Li L, Peng XY. Automated Classification of Inherited Retinal Diseases in Optical Coherence Tomography Images Using Few-shot Learning. Biomedical and environmental sciences : BES. 2023 May 20:36(5):431-440. doi: 10.3967/bes2023.052. Epub [PubMed PMID: 37253669]
Sergouniotis PI, Sohn EH, Li Z, McBain VA, Wright GA, Moore AT, Robson AG, Holder GE, Webster AR. Phenotypic variability in RDH5 retinopathy (Fundus Albipunctatus). Ophthalmology. 2011 Aug:118(8):1661-70. doi: 10.1016/j.ophtha.2010.12.031. Epub 2011 Apr 29 [PubMed PMID: 21529959]
Querques G, Carrillo P, Querques L, Bux AV, Del Curatolo MV, Delle Noci N. High-definition optical coherence tomographic visualization of photoreceptor layer and retinal flecks in fundus albipunctatus associated with cone dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960). 2009 May:127(5):703-6. doi: 10.1001/archophthalmol.2009.87. Epub [PubMed PMID: 19433727]
Level 3 (low-level) evidenceTripathy K, Sarma B, Mazumdar S. Outer retinal tubulation and inner retinal pseudocysts in a patient with maternally inherited diabetes and deafness evaluated with optical coherence tomography angiogram. Indian journal of ophthalmology. 2020 Jan:68(1):250-253. doi: 10.4103/ijo.IJO_577_19. Epub [PubMed PMID: 31856543]
Diaconita V, Kassotis A, Ngo WK. Optical Coherence Tomography Angiography (OCTA) Findings in Retinitis Pigmentosa. Methods in molecular biology (Clifton, N.J.). 2023:2560():101-109. doi: 10.1007/978-1-0716-2651-1_9. Epub [PubMed PMID: 36481887]
Mansour AM, Elnahry AG, Tripathy K, Foster RE, Mehanna CJ, Vishal R, Çavdarlı C, Arrigo A, Parodi MB. Analysis of optical coherence angiography in cystoid macular oedema associated with gyrate atrophy. Eye (London, England). 2021 Jun:35(6):1766-1774. doi: 10.1038/s41433-020-01166-6. Epub 2020 Sep 1 [PubMed PMID: 32873946]
Fleckenstein M, Charbel Issa P, Fuchs HA, Finger RP, Helb HM, Scholl HP, Holz FG. Discrete arcs of increased fundus autofluorescence in retinal dystrophies and functional correlate on microperimetry. Eye (London, England). 2009 Mar:23(3):567-75. doi: 10.1038/eye.2008.59. Epub 2008 Mar 14 [PubMed PMID: 18344954]
Yung M, Klufas MA, Sarraf D. Clinical applications of fundus autofluorescence in retinal disease. International journal of retina and vitreous. 2016:2():12. doi: 10.1186/s40942-016-0035-x. Epub 2016 Apr 8 [PubMed PMID: 27847630]
Casalino G, Khan KN, Armengol M, Wright G, Pontikos N, Georgiou M, Webster AR, Robson AG, Grewal PS, Michaelides M. Autosomal Recessive Bestrophinopathy: Clinical Features, Natural History, and Genetic Findings in Preparation for Clinical Trials. Ophthalmology. 2021 May:128(5):706-718. doi: 10.1016/j.ophtha.2020.10.006. Epub 2020 Oct 8 [PubMed PMID: 33039401]
Naseripour M, Hemmati S, Chaibakhsh S, Gordiz A, Miri L, Abdi F. Cystoid macular oedema without leakage in fluorescein angiography: a literature review. Eye (London, England). 2023 Jun:37(8):1519-1526. doi: 10.1038/s41433-022-02230-z. Epub 2022 Sep 10 [PubMed PMID: 36088420]
Tripathy K. Cystoid Macular Edema in Retinitis Pigmentosa with Intermediate Uveitis Responded Well to Oral and Posterior Subtenon Steroid. Seminars in ophthalmology. 2018:33(4):492-493. doi: 10.1080/08820538.2017.1303521. Epub 2017 Mar 29 [PubMed PMID: 28353369]
Ruia S, Tripathy K. Humphrey Visual Field. StatPearls. 2024 Jan:(): [PubMed PMID: 36256759]
Lee SY, Gurnani B, Mesfin FB. Blindness. StatPearls. 2024 Jan:(): [PubMed PMID: 28846303]
Pinckers A. Berson test for blue cone monochromatism. International ophthalmology. 1992 May:16(3):185-6 [PubMed PMID: 1452423]
Luo X, Cideciyan AV, Iannaccone A, Roman AJ, Ditta LC, Jennings BJ, Yatsenko SA, Sheplock R, Sumaroka A, Swider M, Schwartz SB, Wissinger B, Kohl S, Jacobson SG. Blue cone monochromacy: visual function and efficacy outcome measures for clinical trials. PloS one. 2015:10(4):e0125700. doi: 10.1371/journal.pone.0125700. Epub 2015 Apr 24 [PubMed PMID: 25909963]
Johnson AA, Guziewicz KE, Lee CJ, Kalathur RC, Pulido JS, Marmorstein LY, Marmorstein AD. Bestrophin 1 and retinal disease. Progress in retinal and eye research. 2017 May:58():45-69. doi: 10.1016/j.preteyeres.2017.01.006. Epub 2017 Jan 30 [PubMed PMID: 28153808]
Kortuem FC, Kempf M, Kuehlewein L, Nasser F, Kortuem C, Paques M, Kohl S, Ueffing M, Wissinger B, Zrenner E, Stingl K. Adaptive optics ophthalmoscopy in retinitis pigmentosa (RP): Typical patterns. Acta ophthalmologica. 2022 Nov:100(7):e1539-e1540. doi: 10.1111/aos.15183. Epub 2022 May 25 [PubMed PMID: 35611574]
Shah M, Downes SM, Smithson HE, Young LK. Adaptive optics scanning laser ophthalmoscopy in a heterogenous cohort with Stargardt disease. Scientific reports. 2024 Oct 9:14(1):23629. doi: 10.1038/s41598-024-74088-y. Epub 2024 Oct 9 [PubMed PMID: 39384610]
Britten-Jones AC, Thai L, Flanagan JPM, Bedggood PA, Edwards TL, Metha AB, Ayton LN. Adaptive optics imaging in inherited retinal diseases: A scoping review of the clinical literature. Survey of ophthalmology. 2024 Jan-Feb:69(1):51-66. doi: 10.1016/j.survophthal.2023.09.006. Epub 2023 Sep 29 [PubMed PMID: 37778667]
Level 2 (mid-level) evidenceStrong S, Liew G, Michaelides M. Retinitis pigmentosa-associated cystoid macular oedema: pathogenesis and avenues of intervention. The British journal of ophthalmology. 2017 Jan:101(1):31-37. doi: 10.1136/bjophthalmol-2016-309376. Epub 2016 Dec 2 [PubMed PMID: 27913439]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Schuelke M. Ataxia with Vitamin E Deficiency. GeneReviews(®). 1993:(): [PubMed PMID: 20301419]
Ziccardi L, Cordeddu V, Gaddini L, Matteucci A, Parravano M, Malchiodi-Albedi F, Varano M. Gene Therapy in Retinal Dystrophies. International journal of molecular sciences. 2019 Nov 14:20(22):. doi: 10.3390/ijms20225722. Epub 2019 Nov 14 [PubMed PMID: 31739639]
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, Wittes J, Pappas J, Elci O, McCague S, Cross D, Marshall KA, Walshire J, Kehoe TL, Reichert H, Davis M, Raffini L, George LA, Hudson FP, Dingfield L, Zhu X, Haller JA, Sohn EH, Mahajan VB, Pfeifer W, Weckmann M, Johnson C, Gewaily D, Drack A, Stone E, Wachtel K, Simonelli F, Leroy BP, Wright JF, High KA, Maguire AM. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017 Aug 26:390(10097):849-860. doi: 10.1016/S0140-6736(17)31868-8. Epub 2017 Jul 14 [PubMed PMID: 28712537]
Level 1 (high-level) evidenceBattu R, Ratra D, Gopal L. Newer therapeutic options for inherited retinal diseases: Gene and cell replacement therapy. Indian journal of ophthalmology. 2022 Jul:70(7):2316-2325. doi: 10.4103/ijo.IJO_82_22. Epub [PubMed PMID: 35791112]
Khaboushan AS, Ebadpour N, Moghadam MMJ, Rezaee Z, Kajbafzadeh AM, Zolbin MM. Cell therapy for retinal degenerative disorders: a systematic review and three-level meta-analysis. Journal of translational medicine. 2024 Mar 2:22(1):227. doi: 10.1186/s12967-024-05016-x. Epub 2024 Mar 2 [PubMed PMID: 38431596]
Level 1 (high-level) evidenceFinn AP, Grewal DS, Vajzovic L. Argus II retinal prosthesis system: a review of patient selection criteria, surgical considerations, and post-operative outcomes. Clinical ophthalmology (Auckland, N.Z.). 2018:12():1089-1097. doi: 10.2147/OPTH.S137525. Epub 2018 Jun 13 [PubMed PMID: 29942114]
Le P, Nguyen M, Vu T, Dao DP, Olson D, Zhang AY. Anxiety and Depression in Patients With Retinitis Pigmentosa. Journal of vitreoretinal diseases. 2021 Mar-Apr:5(2):114-120. doi: 10.1177/2474126420936455. Epub 2020 Aug 18 [PubMed PMID: 37009075]
Radu RA, Yuan Q, Hu J, Peng JH, Lloyd M, Nusinowitz S, Bok D, Travis GH. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Investigative ophthalmology & visual science. 2008 Sep:49(9):3821-9. doi: 10.1167/iovs.07-1470. Epub 2008 May 30 [PubMed PMID: 18515570]
Grant CA, Berson EL. Treatable forms of retinitis pigmentosa associated with systemic neurological disorders. International ophthalmology clinics. 2001 Winter:41(1):103-10 [PubMed PMID: 11198137]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Waterham HR, Wanders RJA, Leroy BP. Adult Refsum Disease. GeneReviews(®). 1993:(): [PubMed PMID: 20301527]
Yang JY, Lu B, Feng Q, Alfaro JS, Chen PH, Loscalzo J, Wei WB, Zhang YY, Lu SJ, Wang S. Retinal Protection by Sustained Nanoparticle Delivery of Oncostatin M and Ciliary Neurotrophic Factor Into Rodent Models of Retinal Degeneration. Translational vision science & technology. 2021 Aug 2:10(9):6. doi: 10.1167/tvst.10.9.6. Epub [PubMed PMID: 34347033]
Chen WJ, Ma L, Li MS, Ma X. Valproic acid's effects on visual acuity in retinitis pigmentosa: a systemic review and Meta-analysis. International journal of ophthalmology. 2019:12(1):129-134. doi: 10.18240/ijo.2019.01.20. Epub 2019 Jan 18 [PubMed PMID: 30662852]
Level 1 (high-level) evidenceBrinton GS, Norton EW, Zahn JR, Knighton RW. Ocular quinine toxicity. American journal of ophthalmology. 1980 Sep:90(3):403-10 [PubMed PMID: 7425057]
Level 3 (low-level) evidenceTripathy K, Chawla R, Meena S, Agarwal P. Unilateral giant peripapillary drusen and retinal drusenoid deposits in a case of X-linked retinoschisis. BMJ case reports. 2016 Feb 23:2016():. doi: 10.1136/bcr-2016-214558. Epub 2016 Feb 23 [PubMed PMID: 26907824]
Level 3 (low-level) evidenceBocquet B, El Alami Trebki H, Roux AF, Labesse G, Brabet P, Arndt C, Zanlonghi X, Defoort-Dhellemmes S, Hamroun D, Boulicot-Séguin C, Lequeux L, Picot MC, Huguet H, Audo I, Dhaenens CM, Kalatzis V, Meunier I. Retinitis Punctata Albescens and RLBP1-Allied Phenotypes: Phenotype-Genotype Correlation and Natural History in the Aim of Gene Therapy. Ophthalmology science. 2021 Sep:1(3):100052. doi: 10.1016/j.xops.2021.100052. Epub 2021 Aug 17 [PubMed PMID: 36247817]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, MacDonald IM, Hoang S, Tuupanen S. X-Linked Congenital Stationary Night Blindness. GeneReviews(®). 1993:(): [PubMed PMID: 20301423]
Davies EC, Pineda R 2nd. Cataract surgery outcomes and complications in retinal dystrophy patients. Canadian journal of ophthalmology. Journal canadien d'ophtalmologie. 2017 Dec:52(6):543-547. doi: 10.1016/j.jcjo.2017.04.002. Epub 2017 Jul 22 [PubMed PMID: 29217020]
Yioti G, Stefaniotou M, Ziavrou I, Kotsis K, Hyphantis T. Illness Perceptions, Psychiatric Manifestations, and Quality of Life in Patients with Inherited Retinal Dystrophies. Seminars in ophthalmology. 2017:32(4):428-437. doi: 10.3109/08820538.2015.1118136. Epub 2016 Apr 15 [PubMed PMID: 27082703]
Level 2 (mid-level) evidence