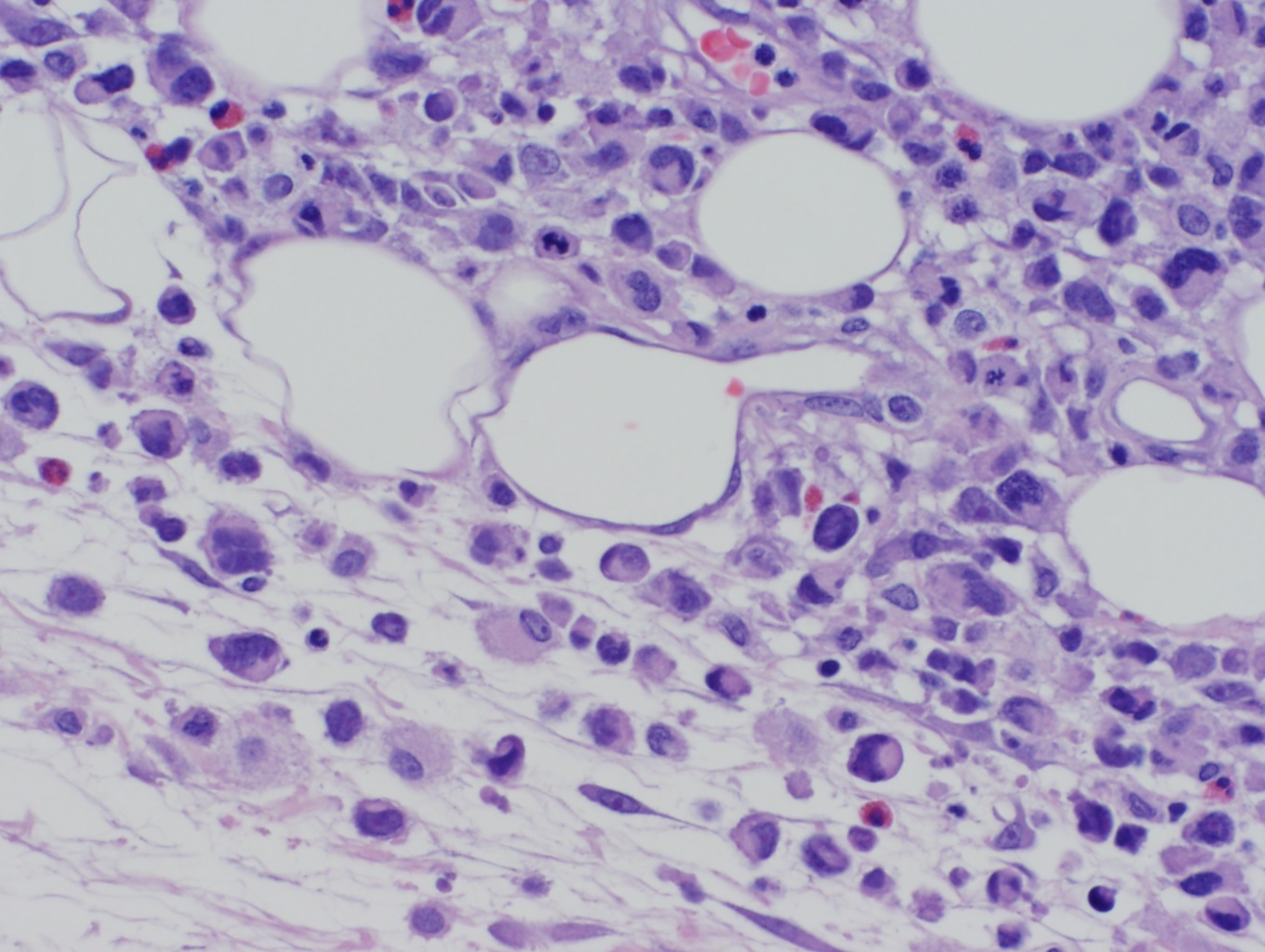
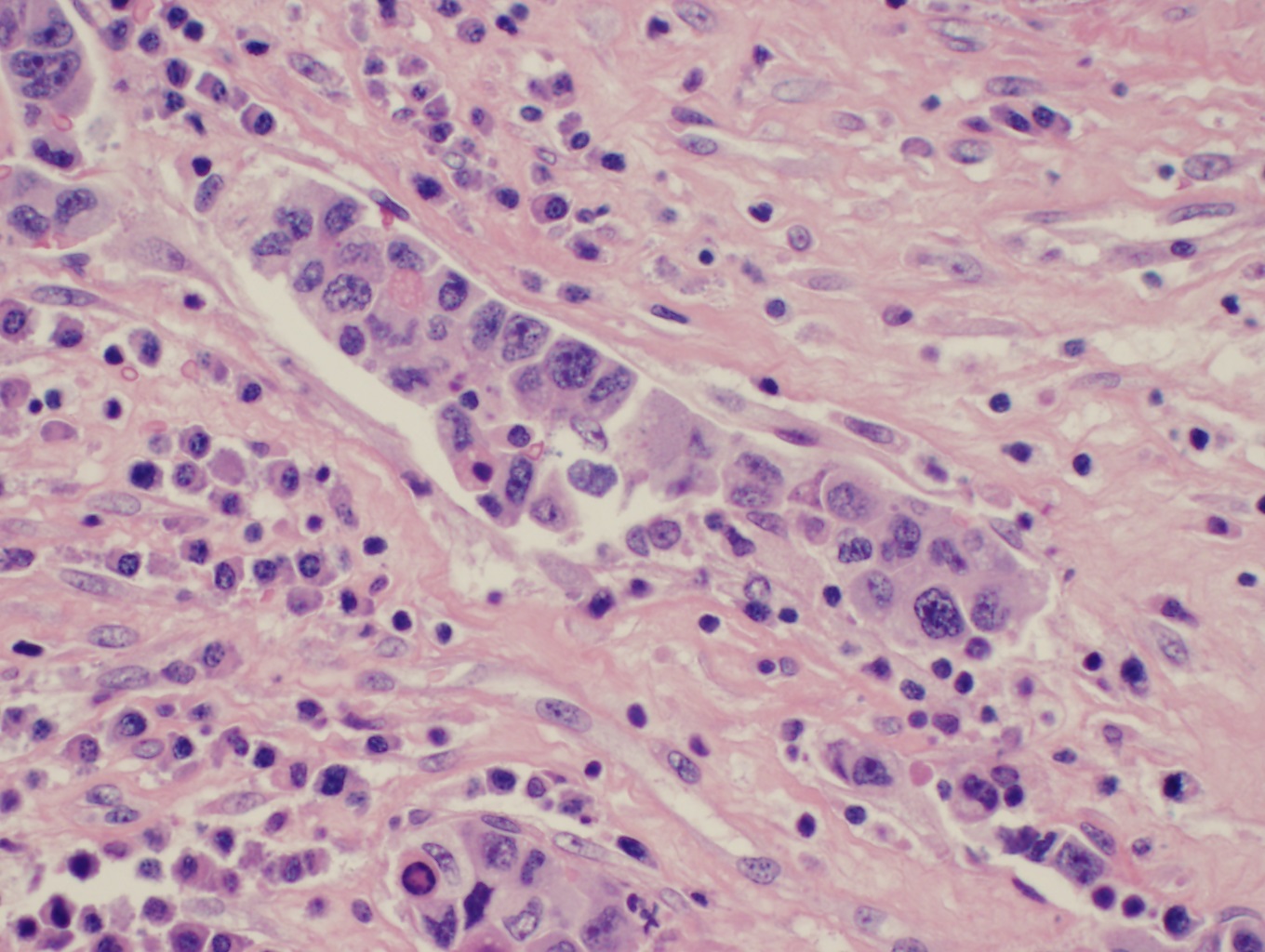
[1]
Ferreri AJ, Govi S, Pileri SA, Savage KJ. Anaplastic large cell lymphoma, ALK-negative. Critical reviews in oncology/hematology. 2013 Feb:85(2):206-15. doi: 10.1016/j.critrevonc.2012.06.004. Epub 2012 Jul 11
[PubMed PMID: 22789917]
[2]
Brody GS. Anaplastic Large Cell Lymphoma Occurring in Women with Breast Implants: Analysis of 173 Cases. Plastic and reconstructive surgery. 2015 Oct:136(4):553e-554e. doi: 10.1097/PRS.0000000000001601. Epub
[PubMed PMID: 26086383]
Level 3 (low-level) evidence
[3]
Jewell M, Spear SL, Largent J, Oefelein MG, Adams WP Jr. Anaplastic large T-cell lymphoma and breast implants: a review of the literature. Plastic and reconstructive surgery. 2011 Sep:128(3):651-661. doi: 10.1097/PRS.0b013e318221db81. Epub
[PubMed PMID: 21865998]
[4]
King RL, Dao LN, McPhail ED, Jaffe ES, Said J, Swerdlow SH, Sattler CA, Ketterling RP, Sidhu JS, Hsi ED, Karikehalli S, Jiang L, Gibson SE, Ondrejka SL, Nicolae A, Macon WR, Dasari S, Parrilla Castellar E, Feldman AL. Morphologic Features of ALK-negative Anaplastic Large Cell Lymphomas With DUSP22 Rearrangements. The American journal of surgical pathology. 2016 Jan:40(1):36-43. doi: 10.1097/PAS.0000000000000500. Epub
[PubMed PMID: 26379151]
[5]
Zeng Y, Feldman AL. Genetics of anaplastic large cell lymphoma. Leukemia & lymphoma. 2016:57(1):21-7. doi: 10.3109/10428194.2015.1064530. Epub 2015 Jul 21
[PubMed PMID: 26104084]
[6]
DeCoteau JF, Butmarc JR, Kinney MC, Kadin ME. The t(2;5) chromosomal translocation is not a common feature of primary cutaneous CD30+ lymphoproliferative disorders: comparison with anaplastic large-cell lymphoma of nodal origin. Blood. 1996 Apr 15:87(8):3437-41
[PubMed PMID: 8605362]
[7]
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016 May 19:127(20):2391-405. doi: 10.1182/blood-2016-03-643544. Epub 2016 Apr 11
[PubMed PMID: 27069254]
[8]
Xing X, Feldman AL. Anaplastic large cell lymphomas: ALK positive, ALK negative, and primary cutaneous. Advances in anatomic pathology. 2015 Jan:22(1):29-49. doi: 10.1097/PAP.0000000000000047. Epub
[PubMed PMID: 25461779]
Level 3 (low-level) evidence
[9]
Grandhi A, Boros AL, Berardo N, Reich RF, Freedman PD. Two cases of CD30+, anaplastic lymphoma kinase (ALK)-negative anaplastic large cell lymphoma with oral manifestations. Oral surgery, oral medicine, oral pathology and oral radiology. 2013 Feb:115(2):e41-7. doi: 10.1016/j.oooo.2012.04.010. Epub 2012 Aug 31
[PubMed PMID: 22940020]
Level 3 (low-level) evidence
[10]
Kodama K, Hokama M, Kawaguchi K, Tanaka Y, Hongo K. Primary ALK-1-negative anaplastic large cell lymphoma of the brain: case report and review of the literature. Neuropathology : official journal of the Japanese Society of Neuropathology. 2009 Apr:29(2):166-71. doi: 10.1111/j.1440-1789.2008.00935.x. Epub 2008 Jun 28
[PubMed PMID: 18564100]
Level 3 (low-level) evidence
[11]
Saikia UN, Sharma N, Duseja A, Bhalla A, Joshi K. Anaplastic large cell lymphoma presenting as acute liver failure: A report of two cases with review of literature. Annals of hepatology. 2010 Oct-Dec:9(4):457-61
[PubMed PMID: 21057166]
Level 3 (low-level) evidence
[12]
Lobo J, Henrique R, Monteiro P, Lobo C. ALK-negative anaplastic large cell lymphoma with urinary bladder involvement diagnosed in urine cytology: A case report and literature review. Diagnostic cytopathology. 2017 Apr:45(4):354-358. doi: 10.1002/dc.23669. Epub 2017 Jan 31
[PubMed PMID: 28139895]
Level 3 (low-level) evidence
[13]
Querfeld C, Khan I, Mahon B, Nelson BP, Rosen ST, Evens AM. Primary cutaneous and systemic anaplastic large cell lymphoma: clinicopathologic aspects and therapeutic options. Oncology (Williston Park, N.Y.). 2010 Jun:24(7):574-87
[PubMed PMID: 20669794]
[14]
Park SH, Chi HS, Cho YU, Jang S, Park CJ. Immunohistopathological features of anaplastic large-cell lymphoma according to anaplastic lymphoma kinase expression and bone marrow involvement pattern. Histopathology. 2013 Jul:63(1):13-8. doi: 10.1111/his.12141. Epub 2013 May 8
[PubMed PMID: 23656317]
[15]
Lu Y, Zhao X, Wang E, Chen W, Huang Q. ALK-negative anaplastic large cell lymphoma with extensive peripheral blood and bone marrow involvements manifested as "leukemic phase". Leukemia research. 2010 Apr:34(4):475-82. doi: 10.1016/j.leukres.2009.07.034. Epub 2009 Aug 19
[PubMed PMID: 19695703]
[16]
Wada DA, Law ME, Hsi ED, Dicaudo DJ, Ma L, Lim MS, Souza Ad, Comfere NI, Weenig RH, Macon WR, Erickson LA, Ozsan N, Ansell SM, Dogan A, Feldman AL. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2011 Apr:24(4):596-605. doi: 10.1038/modpathol.2010.225. Epub 2010 Dec 17
[PubMed PMID: 21169992]
Level 2 (mid-level) evidence
[17]
Herling M, Rassidakis GZ, Jones D, Schmitt-Graeff A, Sarris AH, Medeiros LJ. Absence of Epstein-Barr virus in anaplastic large cell lymphoma: a study of 64 cases classified according to World Health Organization criteria. Human pathology. 2004 Apr:35(4):455-9
[PubMed PMID: 15116326]
Level 3 (low-level) evidence
[18]
Kempf W, Kutzner H, Cozzio A, Sander CA, Pfaltz MC, Müller B, Pfaltz M. MUM1 expression in cutaneous CD30+ lymphoproliferative disorders: a valuable tool for the distinction between lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. The British journal of dermatology. 2008 Jun:158(6):1280-7. doi: 10.1111/j.1365-2133.2008.08566.x. Epub 2008 Apr 10
[PubMed PMID: 18410414]
[19]
Parrilla Castellar ER, Jaffe ES, Said JW, Swerdlow SH, Ketterling RP, Knudson RA, Sidhu JS, Hsi ED, Karikehalli S, Jiang L, Vasmatzis G, Gibson SE, Ondrejka S, Nicolae A, Grogg KL, Allmer C, Ristow KM, Wilson WH, Macon WR, Law ME, Cerhan JR, Habermann TM, Ansell SM, Dogan A, Maurer MJ, Feldman AL. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014 Aug 28:124(9):1473-80. doi: 10.1182/blood-2014-04-571091. Epub 2014 Jun 3
[PubMed PMID: 24894770]
Level 2 (mid-level) evidence
[20]
Fornari A, Piva R, Chiarle R, Novero D, Inghirami G. Anaplastic large cell lymphoma: one or more entities among T-cell lymphoma? Hematological oncology. 2009 Dec:27(4):161-70. doi: 10.1002/hon.897. Epub
[PubMed PMID: 19358142]
[21]
Agnelli L, Mereu E, Pellegrino E, Limongi T, Kwee I, Bergaggio E, Ponzoni M, Zamò A, Iqbal J, Piccaluga PP, Neri A, Chan WC, Pileri S, Bertoni F, Inghirami G, Piva R, European T-Cell Lymphoma Study Group. Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large-cell lymphoma. Blood. 2012 Aug 9:120(6):1274-81
[PubMed PMID: 22740451]
[22]
Al-Rohil RN, Torres-Cabala CA, Patel A, Tetzlaff MT, Ivan D, Nagarajan P, Curry JL, Miranda RN, Duvic M, Prieto VG, Aung PP. Loss of CD30 expression after treatment with brentuximab vedotin in a patient with anaplastic large cell lymphoma: a novel finding. Journal of cutaneous pathology. 2016 Dec:43(12):1161-1166. doi: 10.1111/cup.12797. Epub 2016 Sep 15
[PubMed PMID: 27531242]
[23]
Hudacko R, Rapkiewicz A, Berman RS, Simsir A. ALK-negative anaplastic large cell lymphoma mimicking a soft tissue sarcoma. Journal of cytology. 2011 Oct:28(4):230-3. doi: 10.4103/0970-9371.86362. Epub
[PubMed PMID: 22090705]
[24]
Vij M, Dhir B, Verma R, Agrawal V, Agarwal V, Jaiswal S, Pandey R. Cytomorphology of ALK+ anaplastic large cell lymphoma displaying spindle cells mimicking a sarcomatous tumor: report of a case. Diagnostic cytopathology. 2011 Oct:39(10):775-9. doi: 10.1002/dc.21552. Epub 2010 Oct 1
[PubMed PMID: 20890996]
Level 3 (low-level) evidence
[25]
Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, Rimsza L, Pileri SA, Chhanabhai M, Gascoyne RD, Armitage JO, Weisenburger DD, International Peripheral T-Cell Lymphoma Project. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008 Jun 15:111(12):5496-504. doi: 10.1182/blood-2008-01-134270. Epub 2008 Apr 2
[PubMed PMID: 18385450]
[26]
Zhang Q, Ming J, Zhang S, Li B, Han X, Qiu X. Cytokeratin positivity in anaplastic large cell lymphoma: a potential diagnostic pitfall in misdiagnosis of metastatic carcinoma. International journal of clinical and experimental pathology. 2013:6(4):798-801
[PubMed PMID: 23573330]
Level 2 (mid-level) evidence
[27]
Kinney MC, Collins RD, Greer JP, Whitlock JA, Sioutos N, Kadin ME. A small-cell-predominant variant of primary Ki-1 (CD30)+ T-cell lymphoma. The American journal of surgical pathology. 1993 Sep:17(9):859-68
[PubMed PMID: 8394652]
[28]
Arun I, Roy P, Arora N, Bhave SJ, Nair R, Chandy M. PAX-5 Positivity in Anaplastic Lymphoma Kinase-Negative Anaplastic Large Cell Lymphoma: A Case Report and Review of Literature. International journal of surgical pathology. 2017 Jun:25(4):333-338. doi: 10.1177/1066896916683447. Epub 2016 Dec 26
[PubMed PMID: 28013563]
Level 3 (low-level) evidence
[29]
Venkataraman G, Song JY, Tzankov A, Dirnhofer S, Heinze G, Kohl M, Traverse-Glehen A, Eberle FC, Hanson JC, Raffeld MA, Pittaluga S, Jaffe ES. Aberrant T-cell antigen expression in classical Hodgkin lymphoma is associated with decreased event-free survival and overall survival. Blood. 2013 Mar 7:121(10):1795-804. doi: 10.1182/blood-2012-06-439455. Epub 2013 Jan 10
[PubMed PMID: 23305738]