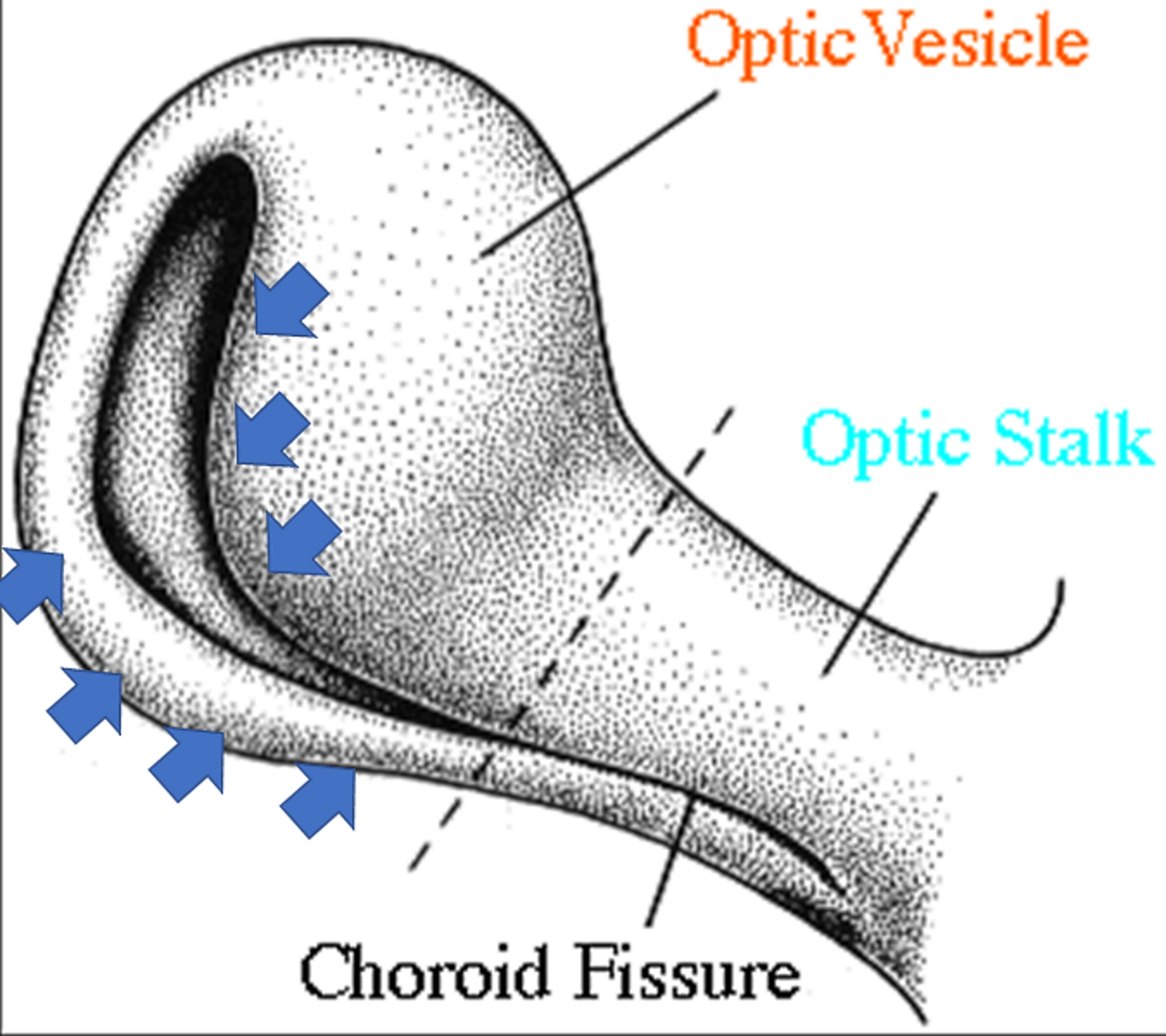
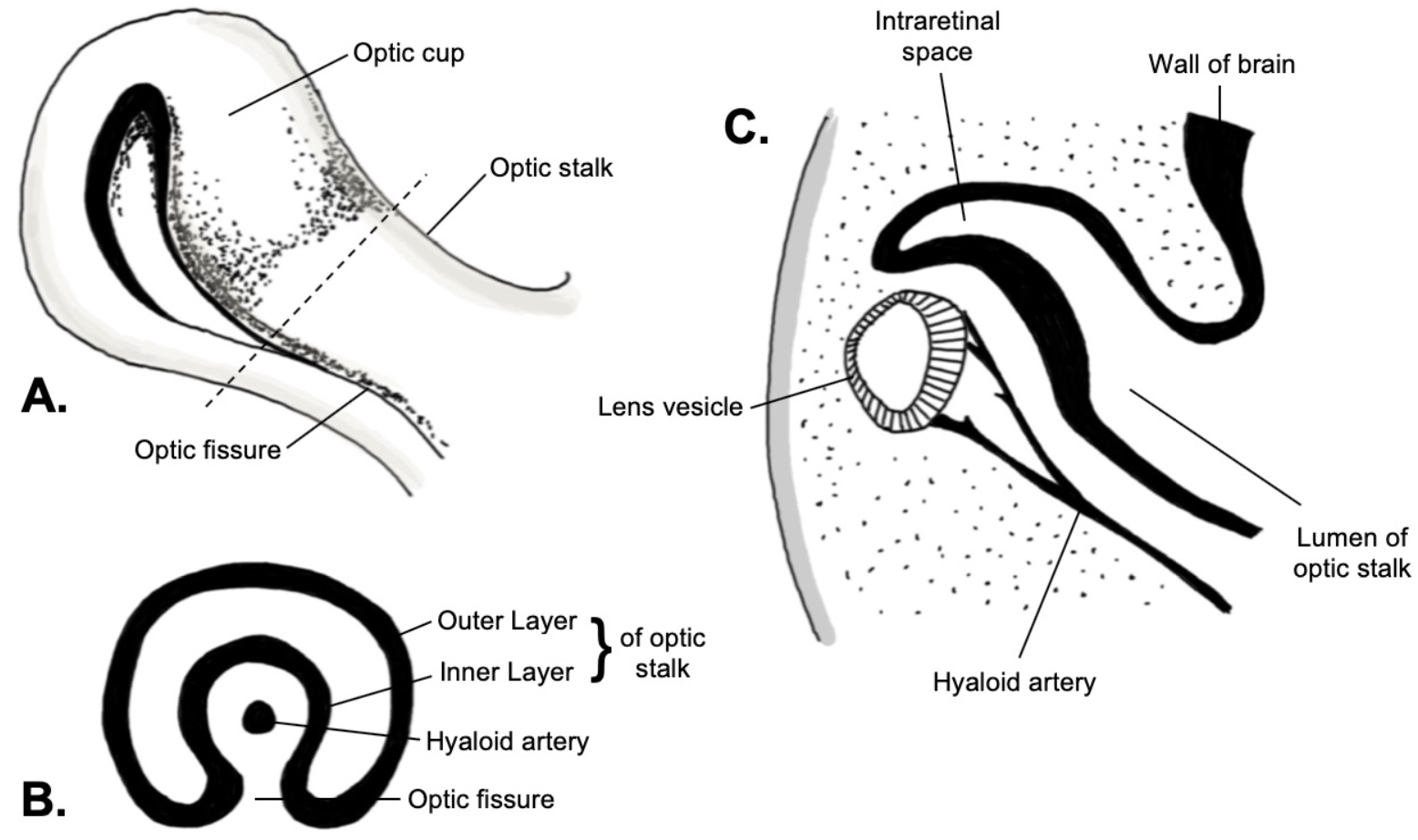
[1]
Eckert P, Knickmeyer MD, Schütz L, Wittbrodt J, Heermann S. Morphogenesis and axis specification occur in parallel during optic cup and optic fissure formation, differentially modulated by BMP and Wnt. Open biology. 2019 Feb 28:9(2):180179. doi: 10.1098/rsob.180179. Epub
[PubMed PMID: 30958096]
[4]
Onwochei BC,Simon JW,Bateman JB,Couture KC,Mir E, Ocular colobomata. Survey of ophthalmology. 2000 Nov-Dec;
[PubMed PMID: 11094243]
Level 3 (low-level) evidence
[5]
Weiss O, Kaufman R, Michaeli N, Inbal A. Abnormal vasculature interferes with optic fissure closure in lmo2 mutant zebrafish embryos. Developmental biology. 2012 Sep 15:369(2):191-8. doi: 10.1016/j.ydbio.2012.06.029. Epub 2012 Jul 20
[PubMed PMID: 22819672]
[6]
Chow RL, Lang RA. Early eye development in vertebrates. Annual review of cell and developmental biology. 2001:17():255-96
[PubMed PMID: 11687490]
[7]
Zagozewski JL, Zhang Q, Eisenstat DD. Genetic regulation of vertebrate eye development. Clinical genetics. 2014 Nov:86(5):453-60. doi: 10.1111/cge.12493. Epub 2014 Sep 25
[PubMed PMID: 25174583]
[8]
Sharma D, Yadav J, Garg E. Cyclopia syndrome. BMJ case reports. 2014 Jun 9:2014():. doi: 10.1136/bcr-2014-203535. Epub 2014 Jun 9
[PubMed PMID: 24913079]
Level 3 (low-level) evidence
[9]
Zhao L, Saitsu H, Sun X, Shiota K, Ishibashi M. Sonic hedgehog is involved in formation of the ventral optic cup by limiting Bmp4 expression to the dorsal domain. Mechanisms of development. 2010 Jan-Feb:127(1-2):62-72. doi: 10.1016/j.mod.2009.10.006. Epub 2009 Oct 23
[PubMed PMID: 19854269]
[10]
Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993 Dec 31:75(7):1417-30
[PubMed PMID: 7916661]
[11]
Zhang L, Mathers PH, Jamrich M. Function of Rx, but not Pax6, is essential for the formation of retinal progenitor cells in mice. Genesis (New York, N.Y. : 2000). 2000 Nov-Dec:28(3-4):135-42
[PubMed PMID: 11105055]
[12]
Behesti H, Papaioannou VE, Sowden JC. Loss of Tbx2 delays optic vesicle invagination leading to small optic cups. Developmental biology. 2009 Sep 15:333(2):360-72. doi: 10.1016/j.ydbio.2009.06.026. Epub 2009 Jul 1
[PubMed PMID: 19576202]
[13]
Wang L, He F, Bu J, Zhen Y, Liu X, Du W, Dong J, Cooney JD, Dubey SK, Shi Y, Gong B, Li J, McBride PF, Jia Y, Lu F, Soltis KA, Lin Y, Namburi P, Liang C, Sundaresan P, Paw BH, Li W, Li DY, Phillips JD, Yang Z. ABCB6 mutations cause ocular coloboma. American journal of human genetics. 2012 Jan 13:90(1):40-8. doi: 10.1016/j.ajhg.2011.11.026. Epub 2012 Jan 5
[PubMed PMID: 22226084]
[14]
Liu C, Widen SA, Williamson KA, Ratnapriya R, Gerth-Kahlert C, Rainger J, Alur RP, Strachan E, Manjunath SH, Balakrishnan A, Floyd JA, UK10K Consortium, Li T, Waskiewicz A, Brooks BP, Lehmann OJ, FitzPatrick DR, Swaroop A. A secreted WNT-ligand-binding domain of FZD5 generated by a frameshift mutation causes autosomal dominant coloboma. Human molecular genetics. 2016 Apr 1:25(7):1382-91. doi: 10.1093/hmg/ddw020. Epub 2016 Jan 24
[PubMed PMID: 26908622]
[15]
Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Verloes A, Drunat S, Pilz D, Di Donato N. Baraitser-Winter Cerebrofrontofacial Syndrome. GeneReviews(®). 1993:():
[PubMed PMID: 26583190]
[16]
Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Current biology : CB. 1995 Aug 1:5(8):931-6
[PubMed PMID: 7583151]
[17]
Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Bardakjian T, Weiss A, Schneider A. Microphthalmia/Anophthalmia/Coloboma Spectrum – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. GeneReviews(®). 1993:():
[PubMed PMID: 20301552]
[18]
Warburg M. Classification of microphthalmos and coloboma. Journal of medical genetics. 1993 Aug:30(8):664-9
[PubMed PMID: 8411053]
[19]
Brown GC, Shields JA, Goldberg RE. Congenital pits of the optic nerve head. II. Clinical studies in humans. Ophthalmology. 1980 Jan:87(1):51-65
[PubMed PMID: 7375086]