Continuing Education Activity
Cerebral cavernous malformations, also known as cavernomas or cavernous hemangiomas, are clusters of abnormal and hyalinized capillaries without intervening brain tissue. Due to recurrent microhemorrhages and thrombosis, they are typically surrounded by hemosiderin deposits and gliosis. These lesions have slow flow and low pressure, causing average rupture risk to be much lower than that of some other vascular malformations such as arteriovenous malformations. Rupture risk is also dependent on the cavernoma's location, presence of associated developmental venous anomaly, and gender. Asymptomatic familial cases are also thought to have a higher annual hemorrhage rate than asymptomatic sporadic cases. Cavernous malformations may also arise de novo. They may grow, shrink, or remain stable over time. This activity reviews the evaluation of cavernous venous malformations and identifies the role of the interprofessional team in managing this condition.
Objectives:
- Review the pathophysiology of cerebral cavernous malformations.
- Describe the workup of a patient with cerebral cavernous malformations.
- Summarize the treatment options for cerebral cavernous malformations.
- Outline the evaluation of cavernous venous malformations and identify the role of the interprofessional team in managing this condition.
Introduction
Cerebral cavernous malformations, also known as cavernomas or cavernous hemangiomas, are clusters of abnormal and hyalinized capillaries without intervening brain tissue. Due to recurrent microhemorrhages and thrombosis, they are typically surrounded by hemosiderin deposits and gliosis. These lesions have slow flow and low pressure, causing average rupture risk to be much lower than that of some other vascular malformations such as arteriovenous malformations. Cavernomas are often found incidentally, but can also present during evaluation of headaches, seizures, focal neurologic deficits, or symptomatic hemorrhage.[1]
Supratentorial location is the most common, though lesions in the basal ganglia, brainstem, cerebellum, and spine also can occur. They can be associated with developmental venous anomalies (DVAs).[2]
The average annual rate of hemorrhage is reported at 0.7%-1.1% per lesion in patients without a history of prior hemorrhage.[3] This risk rises to approximately 4.5% in patients who have sustained previous intracerebral hemorrhage.[3] Approximately 2-3 years after a hemorrhagic event, the risk of hemorrhage is thought to decrease. Rupture risk is also dependent on the cavernoma's location, presence of associated developmental venous anomaly, and gender.[4] Infratentorial location, deep location, young age, and female gender are associated with increased risk.[3],[2] Asymptomatic familial cases are also thought to have a higher annual hemorrhage rate than asymptomatic sporadic cases.[2] Cavernous malformations may also arise de novo.[5] They may grow, shrink, or remain stable over time.[6]
Etiology
Cavernomas can be seen in either familial or sporadic cases. Between 40% to 60% of cases are familial.[1] Sporadic cases tend to present with a single cavernoma; familial cases tend to present with multiple cavernous malformations. In familial cases, an autosomal dominant mutation is usually the culprit in the CCM1/KRIT1, CCM2/Malcavernin, and CCM3/PDCD10 genes located respectfully on the 7q, 7p, and 3p chromosomes.[3][7] Those expressed genes encoded by the CCM genes interact in neural tissue with capillary endothelial tight junctions and cytoskeletal proteins during angiogenesis.[1] A common deletion in CCM2 was found to be responsible for clustering among Ashkenazi Jews.[8]
Recently, genetic studies on the surgically-resected lesions from sporadic cases lacking inherited germline mutations have shown somatic mutations of the same three CCM genes. This could point towards identical molecular mechanisms in both familial and sporadic cavernomas.[9]
Epidemiology
The incidence of cavernomas is approximately 0.4% to 0.8% in the general population.[3] Though uncommon, they are the most common cerebral vascular abnormality and account for 10% to 25% of all vascular malformations.[3] There is no gender predilection.[10] They occur in both adults and children, though the mean patient age is in the fourth decade.[1] The proportion of familial cases of cavernous malformations is reported to be higher in Hispanic-American populations compared with other populations, with rates as high as 50%.[7]
Pathophysiology
Mutations in the cavernoma genes can cause activation of downstream Rho-A kinase (ROCK), which causes endothelial barrier dysfunction. Cavernomas typically most often during the 2nd to 5th decades of life with seizures (50%), symptomatic hemorrhage (25%), and focal neurological deficits without radiographic evidence of recent hemorrhage (25%).[11] Around 20%–50% of patients can have an incidentally detected cavernoma. The risk of first symptomatic hemorrhage has been found to be extremely low (0.08% per patient-year) in the latter population. However, once symptomatic hemorrhage has happened, there is an annual 10-fold increased risk of a subsequent episode of bleeding. This increased risk is highest soon after a hemorrhage but continues thereafter, with a 5-year risk estimated at 42%. Brainstem lesions and CCM3 familial cases with PDCD10/CCM3 mutations are associated with a greater risk of bleeding. Cavernous angiomas with symptomatic hemorrhage (CASH) include lesions that impact a patient’s life and merit clinical intervention.
Histopathology
On histological examination, cavernomas are seen as abnormally dilated and hyalinized capillaries (caverns) without intervening brain tissue. Gliosis and hemosiderin deposits are seen along the margins. Gross pathological appearance is often described as a "mulberry." Light microscopy shows an absence of a smooth muscle wall layer and electron microscopy shows abnormalities in endothelial gap junctions.[12]
History and Physical
Cavernomas can present with headaches, focal neurologic deficits, seizures, hemorrhage, or incidental findings. A review by Ene and colleagues found that the most common presentation of cavernous malformations was seizures at an average of 40.6% of cavernoma cases.[2] Symptomatic hemorrhage has been reported in between 0.7% to 6.5% of cases, although exact rates vary in the literature.[2] A meta-analysis by Washington and colleagues found the most common presentation to be seizures for supratentorial lesions and focal neurologic deficits or ataxia for infratentorial lesions.[10] The proportion of cavernomas that were incidentally diagnosed during an evaluation for unrelated symptoms (headache, for example) has been reported between 6% and 65%.[1] This amount has likely increased over time due to the increasing availability and utilization of magnetic resonance imaging (MRI).[10] History and physical examination of a patient with a newly diagnosed cavernoma should include a thorough review of prior neurologic events, review of any antithrombotic medications, seizure history, detailed neurologic exam, and 3-generation family history, particularly concerning vascular malformations.[13]
Evaluation
Cavernomas can be more difficult to diagnose than other vascular malformations because of their low flow state and small size of the abnormal blood vessels. In particular, cerebral angiography is insensitive for the detection of these lesions, giving rise to the term "angiographically occult".
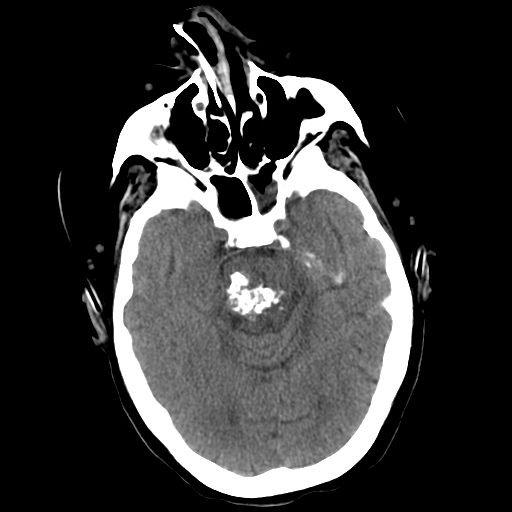
On computed tomography (CT) imaging, cavernomas may appear well demarcated with either a homogeneous or heterogeneous high density related to calcification, thrombosis, increased blood volume, and hemosiderin deposition. They may demonstrate some degree of contrast enhancement. Significant edema or mass effect is unusual except in cases with recent hemorrhage.[14] CT lacks both sensitivity and specificity for cavernous malformations. It should not be the primary imaging study performed in these cases due to the high probability of missing or misdiagnosing lesions.
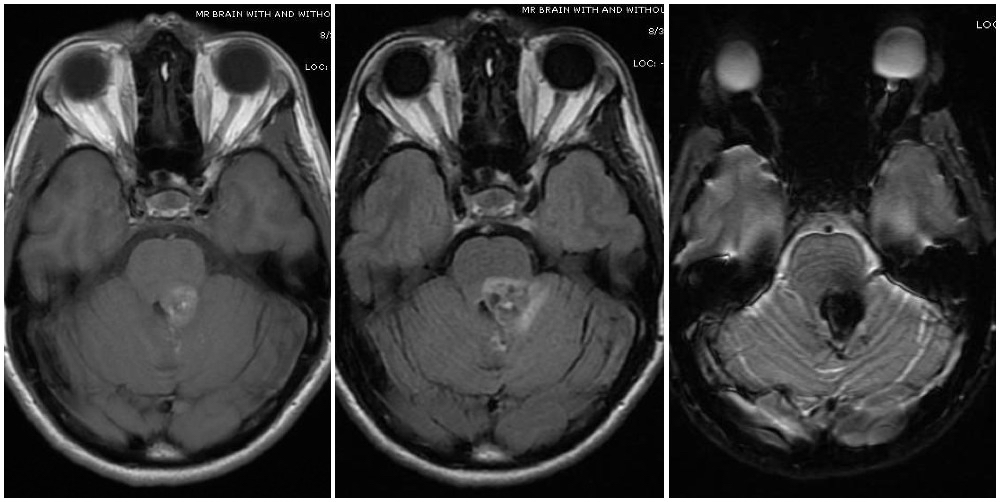
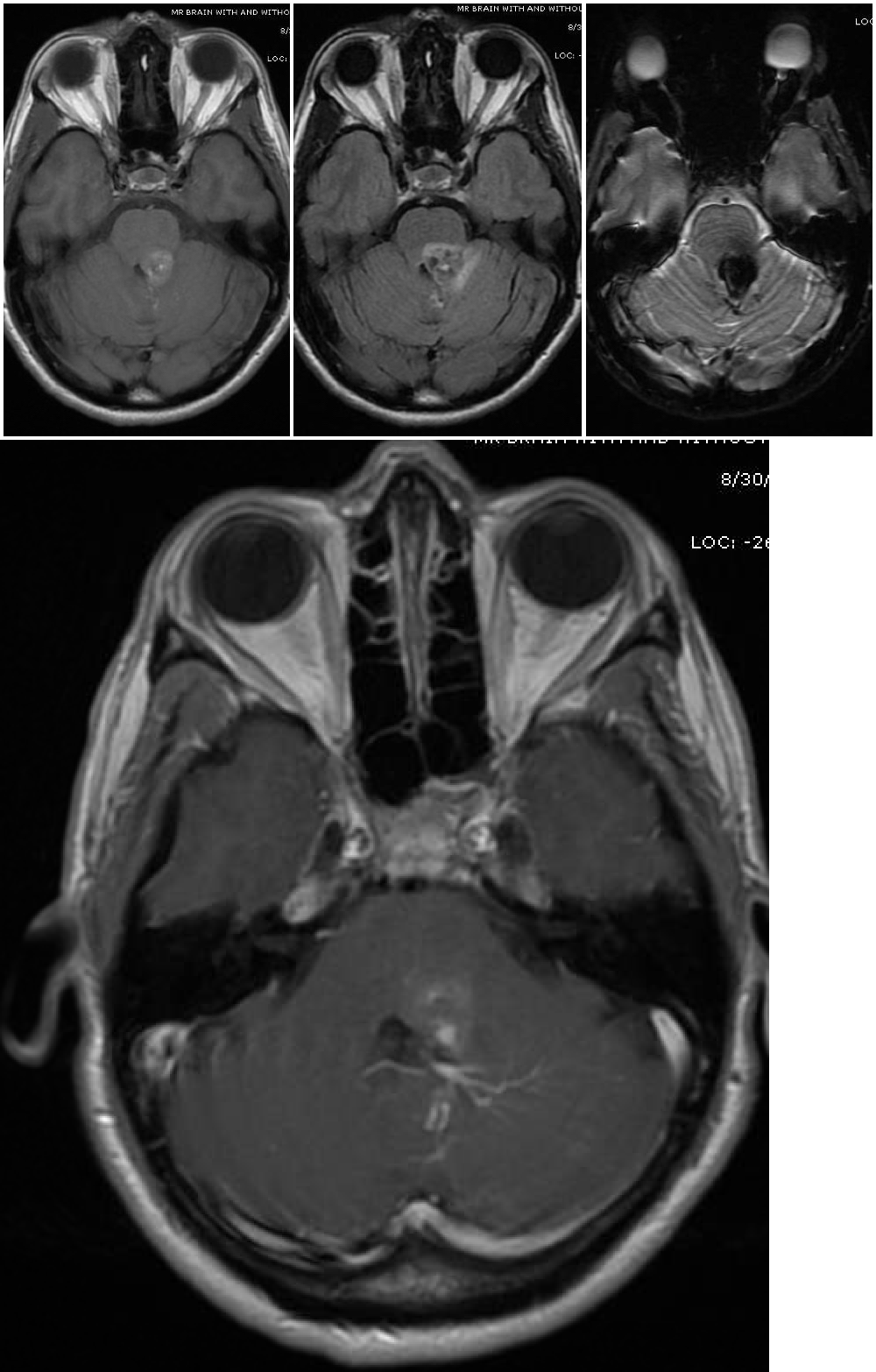
Magnetic resonance imaging (MRI) is the diagnostic modality of choice, particularly gradient-echo sequences (T2*/GRE) showing the "blooming effect of hemosiderin" and susceptibility-weighted imaging sequences (SWI/VenBold) that show areas of prior hemorrhage and multifocal lesions when done at 3T. There are usually mixed signals of blood at different stages of organization and calcifications, with a surrounding hemosiderin ring (best appreciated on T2-weighted sequences).
Cavernous malformations can be divided into four groups (Zabramski classification) based on their appearance on MRI.[15] Type I lesions appear hyperintense on both T1- and T2-weighted sequences due to their subacute hemorrhage and hemosiderin core. Type II lesions represent the classic "popcorn" lesions often described. These type II lesions have mixed signal intensity on both T1 and T2 sequences due to multiple loculated hemorrhages of various stages enclosed by gliotic margins. For these lesions, the gliotic peripheral rim demonstrates decreased signal intensity on T2 sequences. Type III lesions demonstrate chronic resolved hemorrhage within an isointense core. Type IV lesions are thought to represent small capillary telangiectasias that can only be seen on gradient resonance echo sequences. Some authors report an increased risk of hemorrhage for Type 1 and type 2 lesions, prompting consideration for more aggressive surgical management for these types.[1][15] Diffusion tensor sequences and functional MRI (fMRI) can be useful for surgical planning by visualizing white matter tracts and eloquent areas.[3],[1] Presence of an associated developmental venous anomaly on imaging should also be noted due to implications for hemorrhage risk and surgical technique. If several cavernomas surround the periphery of a single developmental venous anomaly, then this is considered part of a single vascular complex consistent with sporadic (versus familial) occurrence.[13]
New MRI techniques - quantitative susceptibility mapping (QSM) and dynamic contrast-enhanced quantitative perfusion (DCEQP) - can measure the quantity of iron deposition and vascular permeability in cavernomas and can be potential biomarkers of disease activity.[16]
Treatment / Management
Conservative management with serial imaging and observation, microsurgical resection, and stereotactic radiosurgery are the 3 main options for the management of cerebral cavernomas. When discussing treatment options, the risks of intervention versus the risks of having an untreated cavernous malformation must be weighed carefully. The natural history of cavernous malformations, clinical presentation, lesion location, number of hemorrhagic events, and medical comorbidities help guide this discussion.[17] For small and incidentally discovered lesions, observation is often reasonable.[17]
According to the Cerebral Cavernous Malformations Care Guidelines published in 2017 by the Angioma Alliance (https://www.angioma.org/for-professionals/clinicians/ccm-care-guidelines), surgical resection is not recommended for asymptomatic cavernous malformations. This is particularly true for multiple lesions or situations in which the lesions are located in deep, eloquent, or brainstem tissue. Surgical resection may be considered for a solitary asymptomatic cavernous malformation if the lesion is in a non-eloquent and accessible location. In those cases, the benefits to resection may include prevention of hemorrhage, reduction of time and costs of follow-up, relief of psychological burden, lifestyle or career decisions, or facilitation of future anti-coagulation if needed.[13]
As per those same guidelines, early surgical resection should be considered for medically refractory epilepsy when the epileptogenic focus is related to the cavernoma. Such early intervention may prevent a "kindling" effect and improve the likelihood of seizure freedom. For symptomatic and easily accessible cavernous hemangiomas, it has been suggested that morbidity and mortality associated with surgery are approximately equivalent to the risk of living with a cavernoma for 2 years. For symptomatic and deeply located cavernomas, morbidity and mortality associated with surgical resection may be considered to be approximately equivalent to living with the cavernoma for 5 to 10 years. While surgery is not typically recommended for brainstem cavernomas due to a significant risk of complications, many recommend operating after a second symptomatic bleed due to the concept that those cavernomas may have a more aggressive natural course. Indications for resection of brainstem cavernomas after a single disabling hemorrhage or spinal cavernomas are more controversial.[13]
For cases undergoing surgical resection, steroids may be considered 1 to 2 weeks before surgery to reduce edema and aid with resection. Resection of any associated developmental venous anomalies should be strictly avoided due to potential venous infarction. Complete removal of the lesion is required to prevent recurrent hemorrhages. If the surgery is being performed for seizures, the hemosiderin-stained surrounding neural parenchyma and gliosis should also be resected when feasible. After surgery, MRI within 72 hours is recommended to ensure complete resection. Some authors report a 40% risk of rebleeding of cavernoma remnants after partial resection.[3]
Stereotactic radiosurgery has been proposed as an alternative to microsurgery for treatment, although the effect of radiosurgery on reducing risks of re-bleeding remains controversial. Even for proponents of the technique, radiosurgery is thought to have a 2-year latency period before optimal risk reduction of recurrent hemorrhage occurs. While microsurgical resection remains the standard treatment for cavernomas, stereotactic radiosurgery is an option for solitary symptomatic lesions with an unacceptably high surgical risk.[3][13] In general, it is not recommended for cavernous malformations that are asymptomatic, surgically accessible, or familial due to the risk of de novo cavernoma formation.[13]
For patients with a newly diagnosed cerebral cavernoma and with no history of brain radiation, no positive family history, or multiple cavernous malformations, genetic testing should be considered. If a positive mutation is found, genetic counseling should be offered to the patient and family.[13] First-degree relatives of patients with more than one family member with a cavernoma should also have a screening MRI recommended as well as genetic counseling.
Medical treatment (still in trial stage) with ROCK inhibitor, statins (with multiple effects including Rho inhibition), vitamin D, and B-cell depletion has shown to decrease hemorrhage in cavernomas.[18]
Differential Diagnosis
Differential diagnosis may include other cerebral vascular malformations such as arteriovenous malformations, venous angiomas, capillary telangiectasias, dural arteriovenous fistulas, aneurysm, and vein of Galen malformations. MRI is the best study to diagnose cerebral cavernous malformations due to characteristic imaging findings, although angiography for the exclusion of other cerebrovascular pathologies under consideration may be helpful in certain clinical settings (such as spontaneous intracranial hemorrhage).
Prognosis
Prognosis depends on the location of the bleed and the degree of neurological deficits.
Complications
The main complications associated with cavernomas include intracerebral hemorrhage, neurological deficits, seizures, and rarely a risk to life.
Consultations
Neurosurgery consultation is recommended for the management of patients with a cerebral or spinal cavernous malformation.
Deterrence and Patient Education
Some activities might have to be restricted for people with cavernomas (not proven), which include mountain climbing above 10000 feet, smoking, water activities, and contact sports. Most studies suggest that the use of antiplatelet medications (for other conditions, if needed) is safe in these patients.
Enhancing Healthcare Team Outcomes
MRI is the standard imaging modality for the diagnosis of cavernous malformations. Cerebral angiography is typically not indicated unless exclusion of other cerebrovascular pathology is being considered.
- For an initial seizure thought to be related to a cavernoma, anti-epileptic medication is a reasonable first-line therapy (Class I, Level B). Surgical resection should be considered for medically refractory epilepsy when the epileptogenic focus is related to a cavernoma.[13]
- Genetic counseling should be considered for those with familial or multiple cavernomas (Class I, Level C).[13]
- Neurosurgical consultation is indicated for the management of cavernous malformations. Treatment options include observation with serial imaging, surgical resection, and stereotactic radiosurgery.