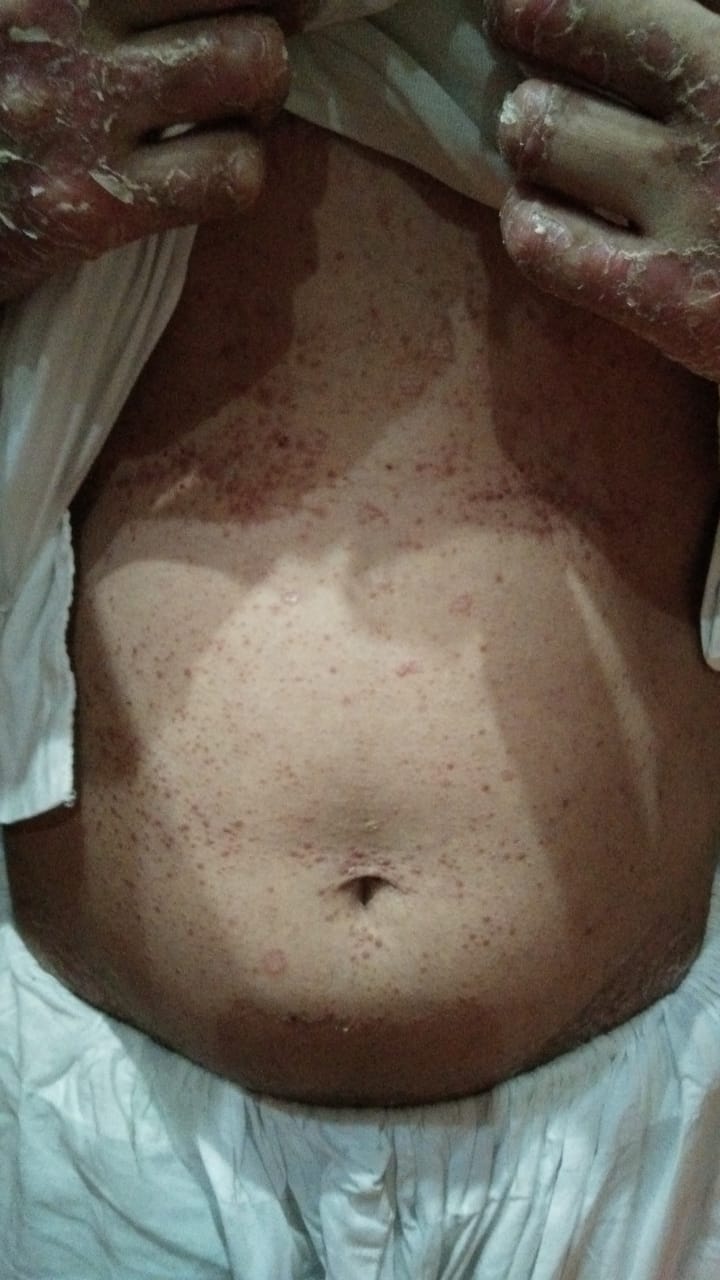
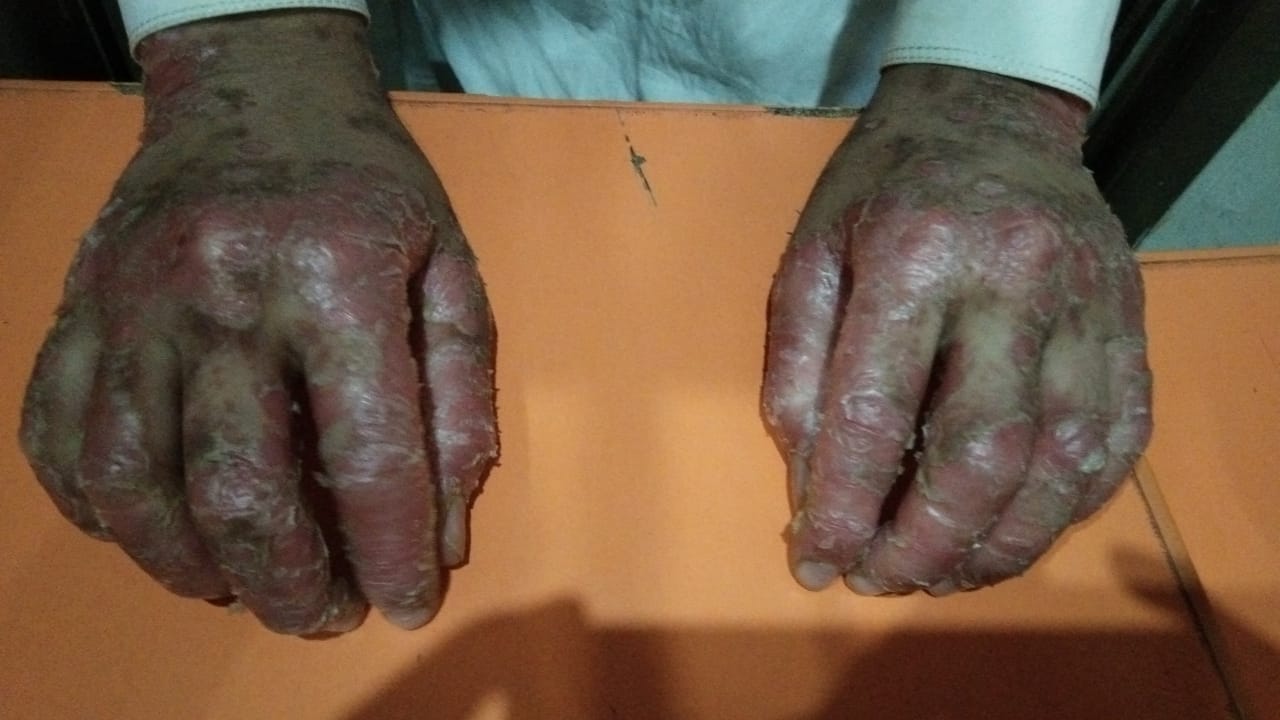
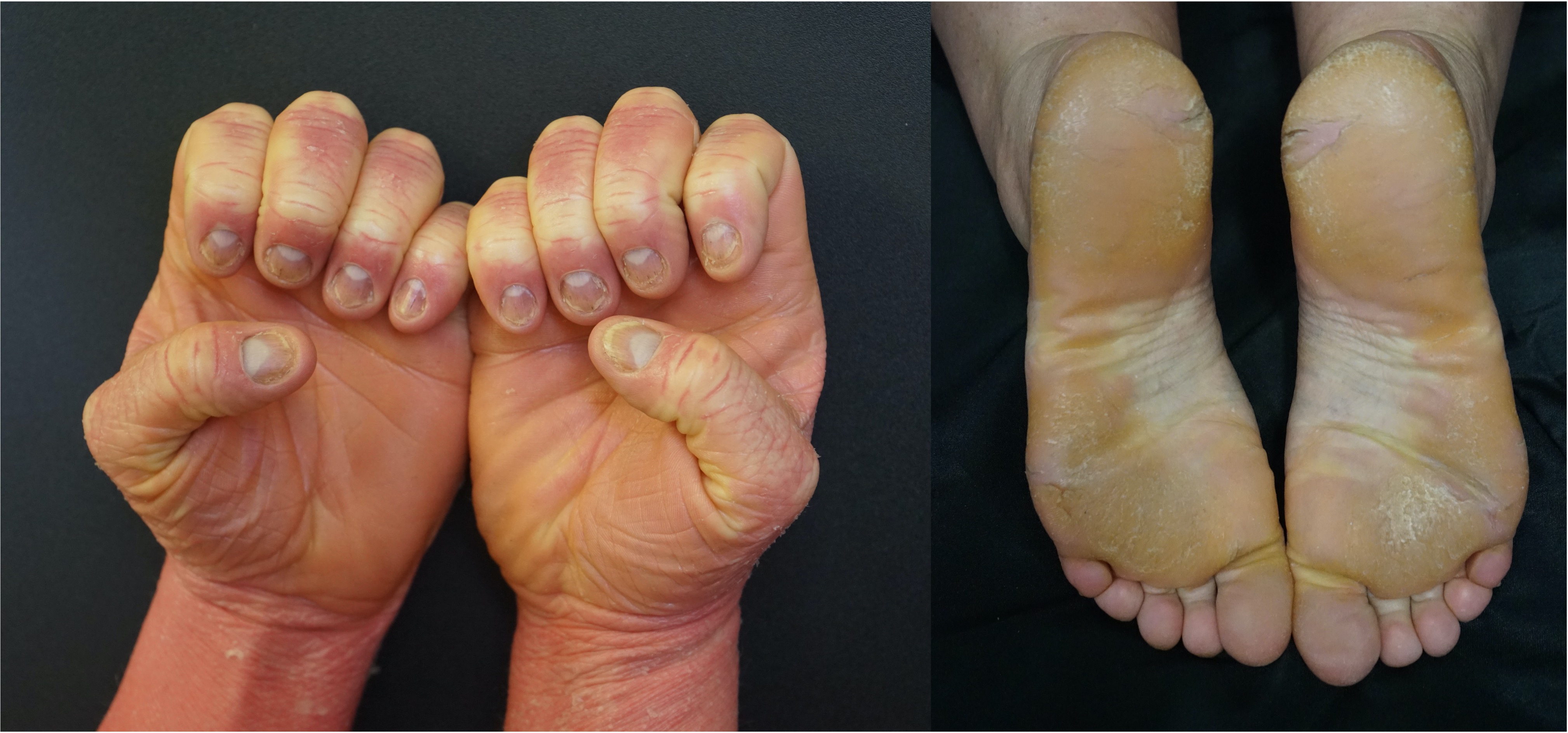
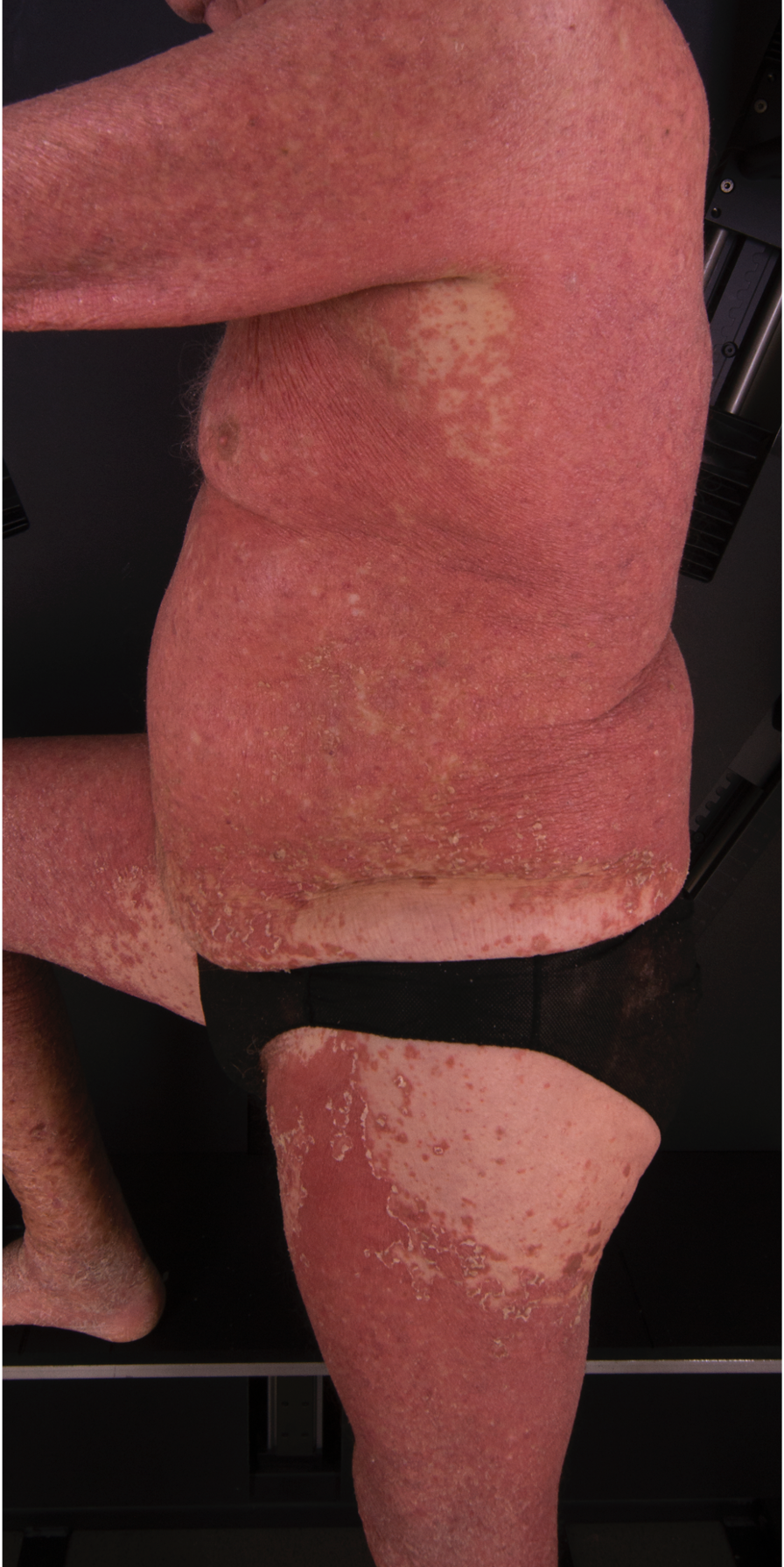
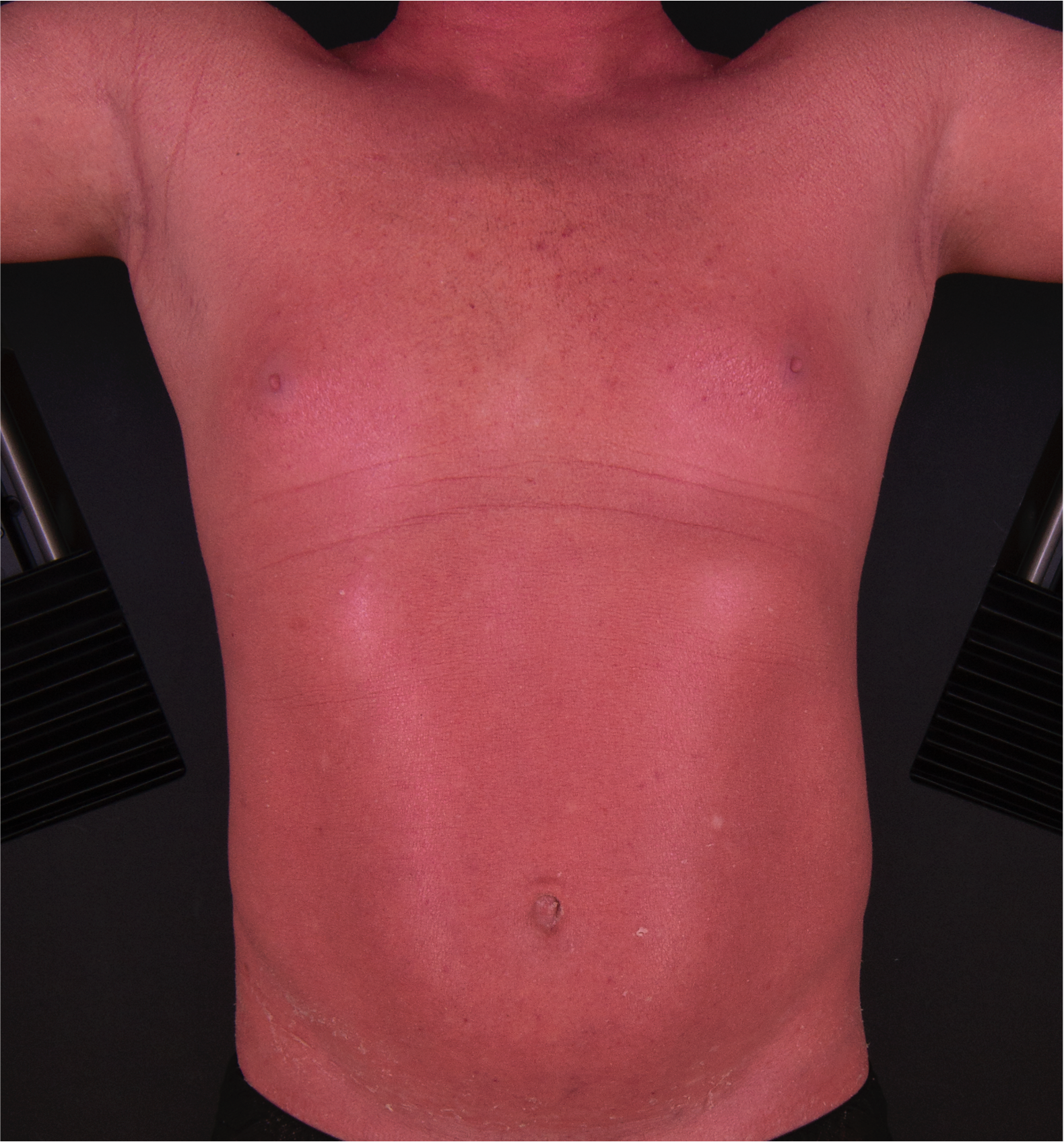
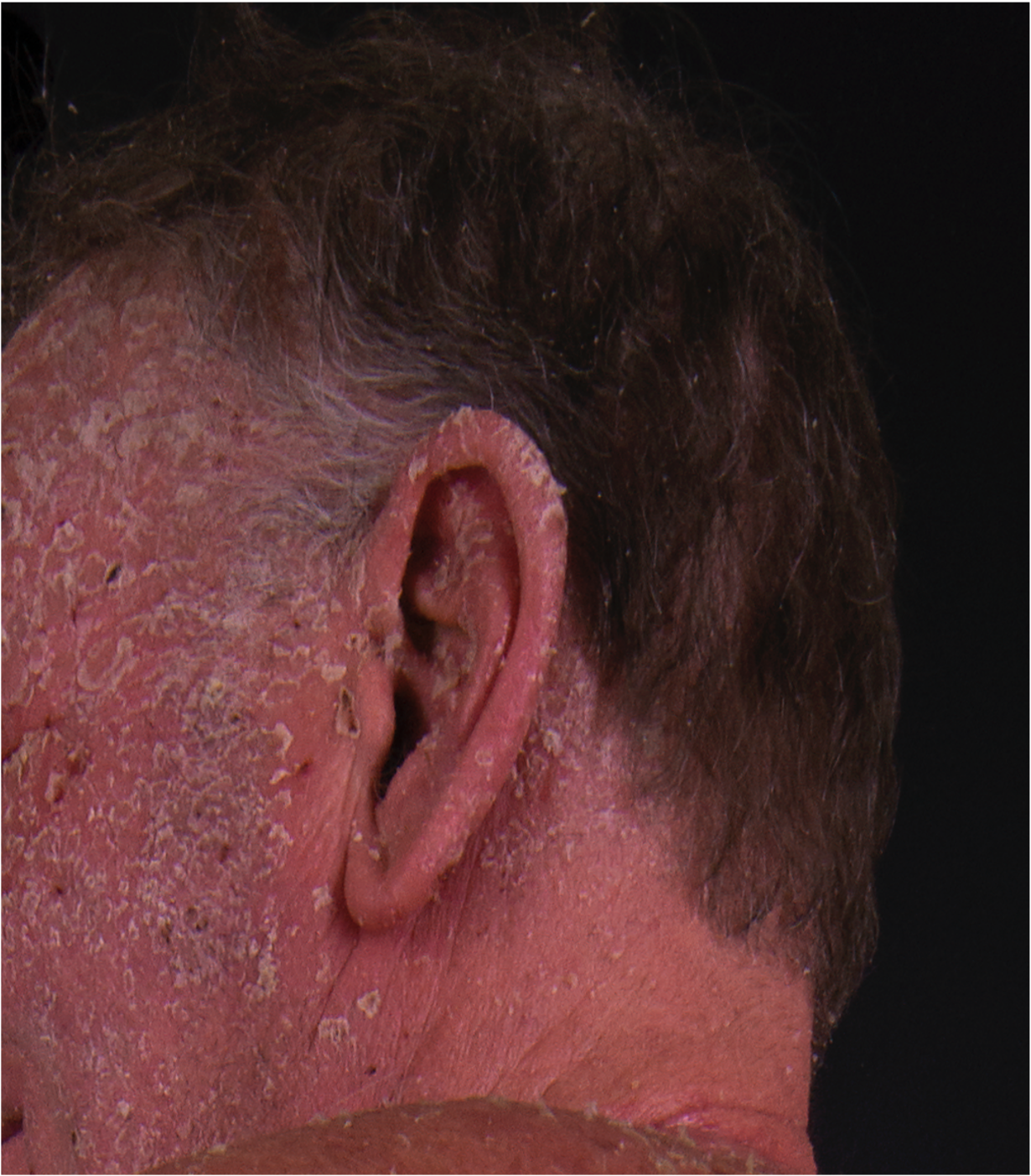
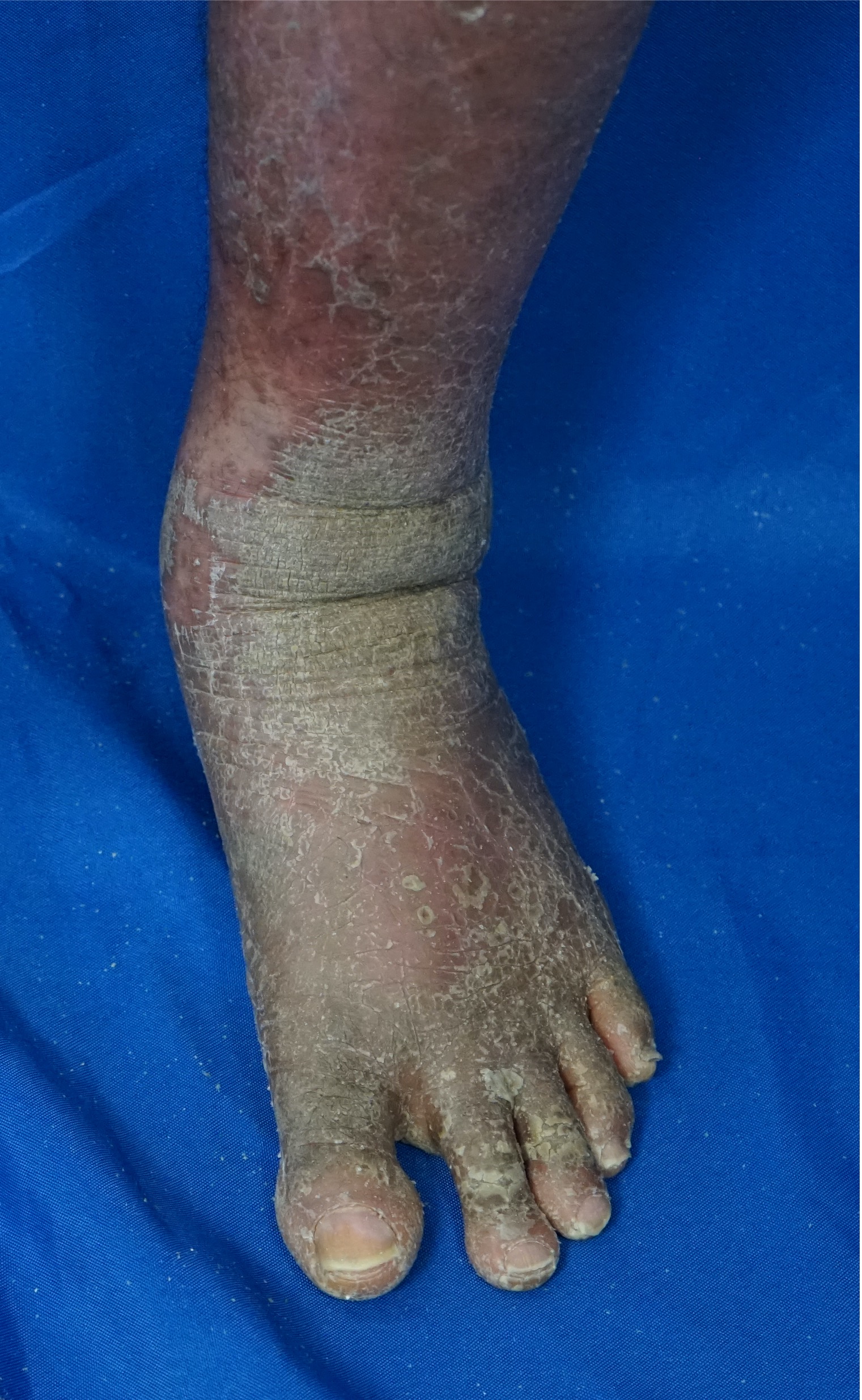
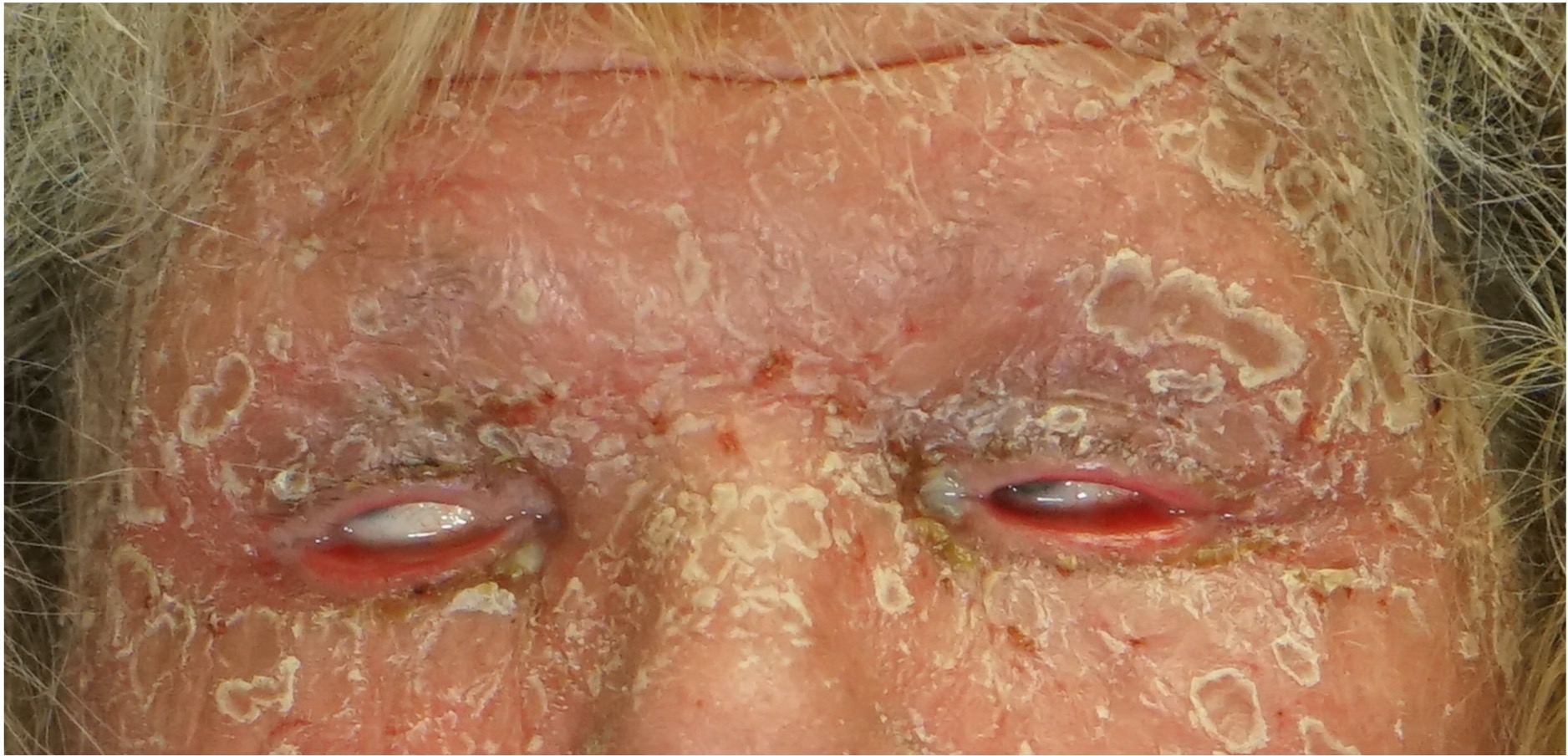
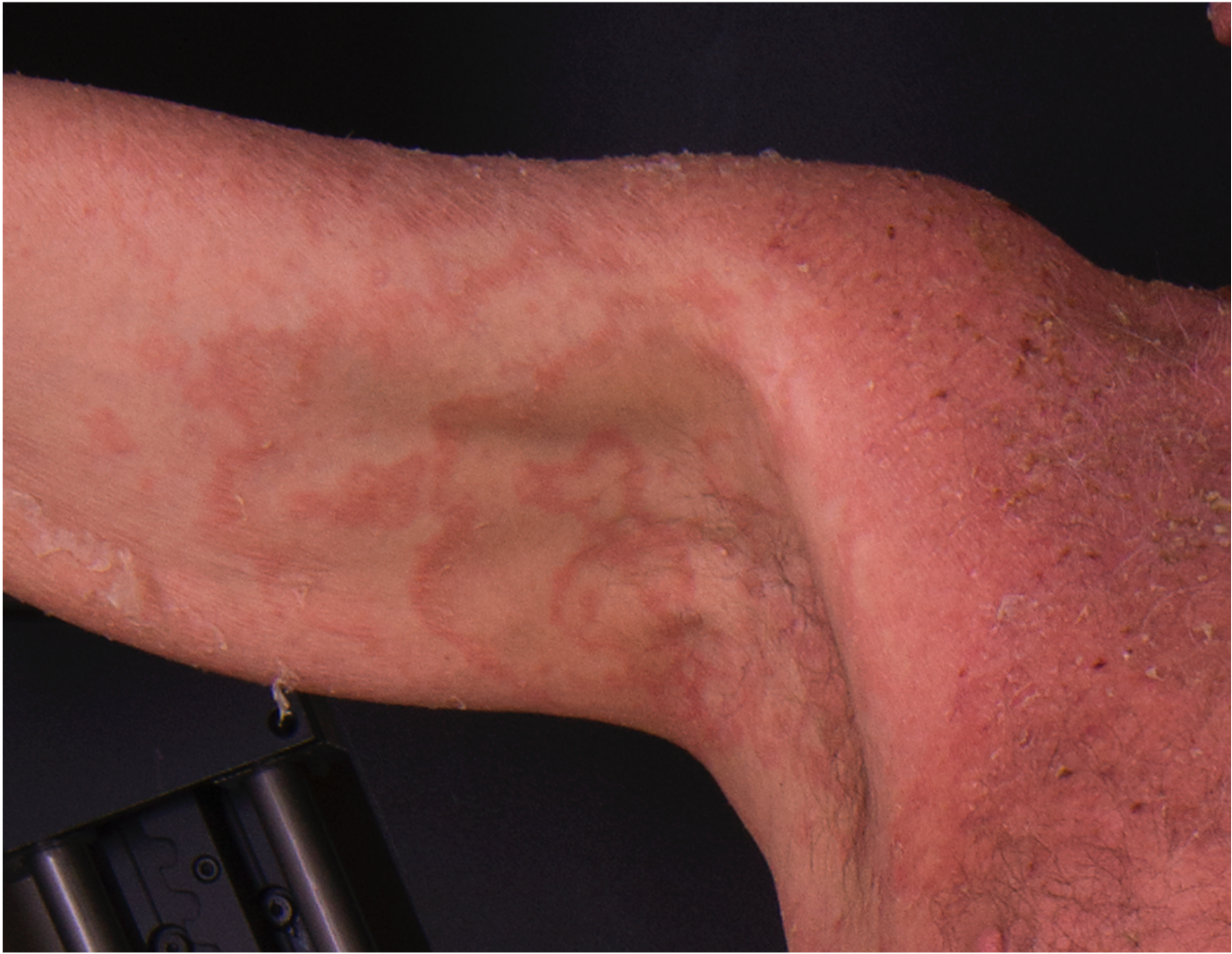
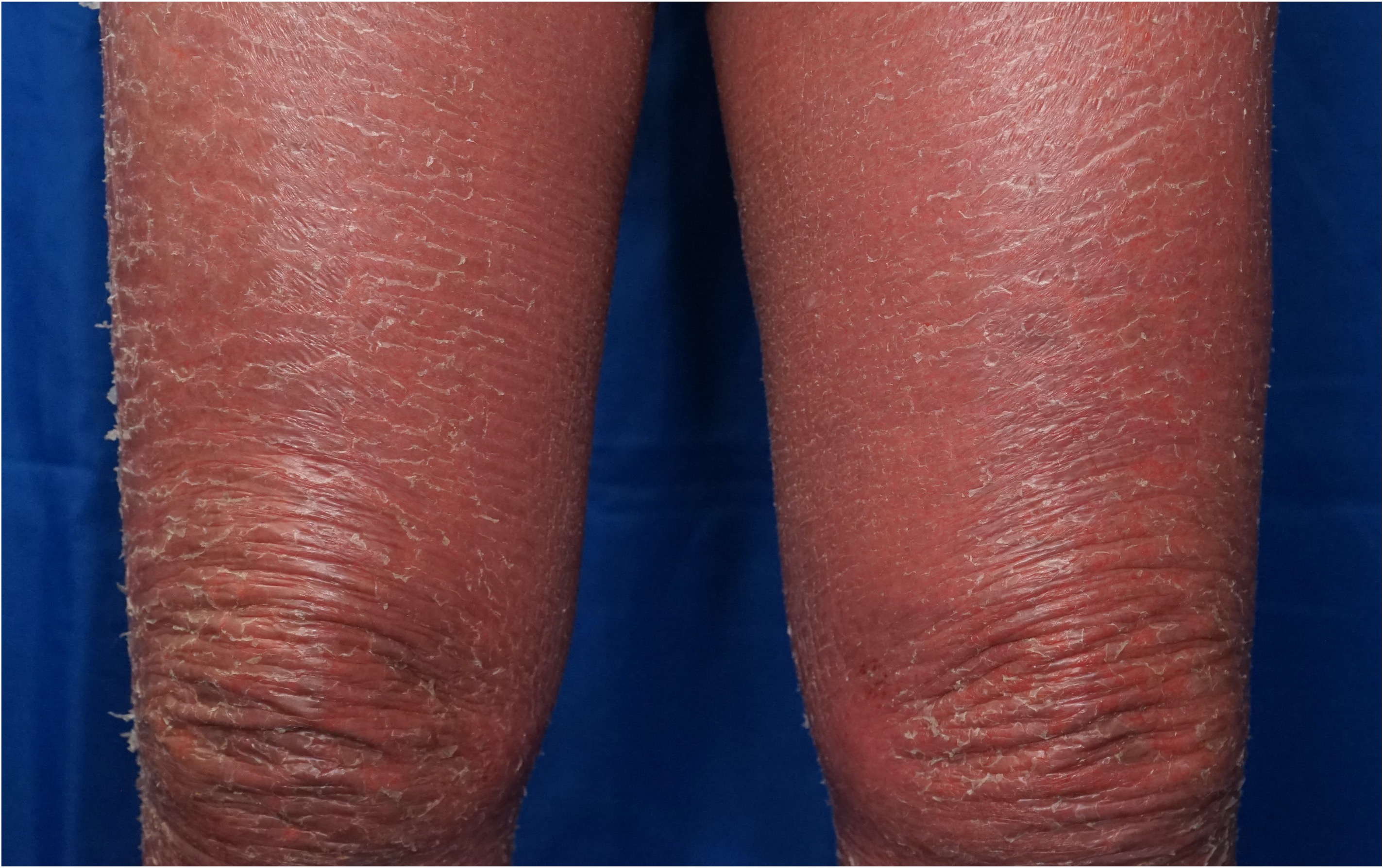
[2]
Eastham AB. Pityriasis Rubra Pilaris. JAMA dermatology. 2019 Mar 1:155(3):404. doi: 10.1001/jamadermatol.2018.5030. Epub
[PubMed PMID: 30725099]
[3]
Eastham AB, Tkachenko EY, Femia AN, Pappas-Taffer LK, Rosenbach M, Joyce CJ, Liu S, Vleugels RA. Pityriasis rubra pilaris: A study evaluating patient quality of life in 2 populations. Journal of the American Academy of Dermatology. 2019 Aug:81(2):638-640. doi: 10.1016/j.jaad.2019.01.061. Epub 2019 Jan 30
[PubMed PMID: 30710600]
Level 2 (mid-level) evidence
[4]
Gál B, Göblös A, Danis J, Farkas K, Sulák A, Varga E, Nagy N, Széll M, Kemény L, Bata-Csörgő Z. The management and genetic background of pityriasis rubra pilaris: a single-centre experience. Journal of the European Academy of Dermatology and Venereology : JEADV. 2019 May:33(5):944-949. doi: 10.1111/jdv.15455. Epub 2019 Mar 3
[PubMed PMID: 30697821]
[5]
Cribier B. [The very first images in the Annales de dermatologie et de syphiligraphie, 1868-1889]. Annales de dermatologie et de venereologie. 2018 Dec:145 Suppl 6():VIS63-VIS100. doi: 10.1016/S0151-9638(18)31295-X. Epub
[PubMed PMID: 30598136]
[6]
Mufti A, Lytvyn Y, Maliyar K, Sachdeva M, Yeung J. Drugs associated with development of pityriasis rubra pilaris: A systematic review. Journal of the American Academy of Dermatology. 2021 Apr:84(4):1071-1081. doi: 10.1016/j.jaad.2020.07.052. Epub 2020 Jul 17
[PubMed PMID: 32687965]
Level 1 (high-level) evidence
[7]
Joshi TP, Duvic M. Pityriasis Rubra Pilaris: An Updated Review of Clinical Presentation, Etiopathogenesis, and Treatment Options. American journal of clinical dermatology. 2024 Mar:25(2):243-259. doi: 10.1007/s40257-023-00836-x. Epub 2023 Dec 30
[PubMed PMID: 38159213]
[8]
Sehgal VN, Jain MK, Mathur RP. Pityriasis rubra pilaris in Indians. The British journal of dermatology. 1989 Dec:121(6):821-2
[PubMed PMID: 2611132]
[9]
Verhagen AR, Koten JW, Chaddah VK, Patel RI. Skin diseases in Kenya. A clinical and histopathological study of 3,168 patients. Archives of dermatology. 1968 Dec:98(6):577-86
[PubMed PMID: 4235165]
[11]
Davidson CL Jr, Winkelmann RK, Kierland RR. Pityriasis rubra pilaris. A follow-up study of 57 patients. Archives of dermatology. 1969 Aug:100(2):175-8
[PubMed PMID: 5797957]
[12]
Fuchs-Telem D, Sarig O, van Steensel MA, Isakov O, Israeli S, Nousbeck J, Richard K, Winnepenninckx V, Vernooij M, Shomron N, Uitto J, Fleckman P, Richard G, Sprecher E. Familial pityriasis rubra pilaris is caused by mutations in CARD14. American journal of human genetics. 2012 Jul 13:91(1):163-70. doi: 10.1016/j.ajhg.2012.05.010. Epub 2012 Jun 14
[PubMed PMID: 22703878]
[13]
Li Q, Jin Chung H, Ross N, Keller M, Andrews J, Kingman J, Sarig O, Fuchs-Telem D, Sprecher E, Uitto J. Analysis of CARD14 Polymorphisms in Pityriasis Rubra Pilaris: Activation of NF-κB. The Journal of investigative dermatology. 2015 Jul:135(7):1905-1908. doi: 10.1038/jid.2015.65. Epub 2015 Mar 3
[PubMed PMID: 25734815]
[14]
Jordan CT, Cao L, Roberson ED, Pierson KC, Yang CF, Joyce CE, Ryan C, Duan S, Helms CA, Liu Y, Chen Y, McBride AA, Hwu WL, Wu JY, Chen YT, Menter A, Goldbach-Mansky R, Lowes MA, Bowcock AM. PSORS2 is due to mutations in CARD14. American journal of human genetics. 2012 May 4:90(5):784-95. doi: 10.1016/j.ajhg.2012.03.012. Epub 2012 Apr 19
[PubMed PMID: 22521418]
[15]
Haynes D, Reiter T, Velasco R, Chang M, Kulkarni R, Kent G, Strunck J, Cassidy P, Greiling TM. Pityriasis Rubra Pilaris Transcriptomics Implicate T Helper 17 Signaling and Correlate with Response to Ixekizumab, with Distinct Gene Expression Profiles in Nonresponders. The Journal of investigative dermatology. 2023 Mar:143(3):501-504.e1. doi: 10.1016/j.jid.2022.09.005. Epub 2022 Sep 24
[PubMed PMID: 36167251]
[16]
Strunck JL, Cutler B, Rajpal B, Kent G, Haynes D, Topham CA, Ortega-Loayza AG, Yang D, Wang Z, Liu Y, Cassidy P, Greiling TM. Pityriasis Rubra Pilaris Response to IL-17A Inhibition Is Associated with IL-17C and CCL20 Protein Levels. The Journal of investigative dermatology. 2022 Jan:142(1):235-239.e1. doi: 10.1016/j.jid.2021.06.009. Epub 2021 Jul 9
[PubMed PMID: 34246621]
[17]
Mellett M, Meier B, Mohanan D, Schairer R, Cheng P, Satoh TK, Kiefer B, Ospelt C, Nobbe S, Thome M, Contassot E, French LE. CARD14 Gain-of-Function Mutation Alone Is Sufficient to Drive IL-23/IL-17-Mediated Psoriasiform Skin Inflammation In Vivo. The Journal of investigative dermatology. 2018 Sep:138(9):2010-2023. doi: 10.1016/j.jid.2018.03.1525. Epub 2018 Apr 22
[PubMed PMID: 29689250]
[18]
Wang M, Zhang S, Zheng G, Huang J, Songyang Z, Zhao X, Lin X. Gain-of-Function Mutation of Card14 Leads to Spontaneous Psoriasis-like Skin Inflammation through Enhanced Keratinocyte Response to IL-17A. Immunity. 2018 Jul 17:49(1):66-79.e5. doi: 10.1016/j.immuni.2018.05.012. Epub 2018 Jul 3
[PubMed PMID: 29980436]
[19]
Shao S, Chen J, Swindell WR, Tsoi LC, Xing X, Ma F, Uppala R, Sarkar MK, Plazyo O, Billi AC, Wasikowski R, Smith KM, Honore P, Scott VE, Maverakis E, Kahlenberg JM, Wang G, Ward NL, Harms PW, Gudjonsson JE. Phospholipase A2 enzymes represent a shared pathogenic pathway in psoriasis and pityriasis rubra pilaris. JCI insight. 2021 Oct 22:6(20):. pii: e151911. doi: 10.1172/jci.insight.151911. Epub 2021 Oct 22
[PubMed PMID: 34491907]
[20]
Ghatnekar S, Shah N, Min MS, Mazori DR, LaChance AH, Vleugels RA, Nambudiri VE. Clinical features and eosinophilia in pityriasis rubra pilaris: A multicenter cohort. Journal of the American Academy of Dermatology. 2022 Apr:86(4):907-909. doi: 10.1016/j.jaad.2021.03.043. Epub 2021 Mar 24
[PubMed PMID: 33771592]
[21]
Avitan-Hersh E, Bergman R. The Incidence of Acantholysis in Pityriasis Rubra Pilaris-Histopathological Study Using Multiple-Step Sections and Clinicopathologic Correlations. The American Journal of dermatopathology. 2015 Oct:37(10):755-8. doi: 10.1097/DAD.0000000000000346. Epub
[PubMed PMID: 26381023]
[22]
Haynes D, Strunck JL, Topham CA, Ortega-Loayza AG, Kent G, Cassidy PB, Hu R, Choate K, Wang Z, Liu Y, Greiling TM. Evaluation of Ixekizumab Treatment for Patients With Pityriasis Rubra Pilaris: A Single-Arm Trial. JAMA dermatology. 2020 Jun 1:156(6):668-675. doi: 10.1001/jamadermatol.2020.0932. Epub
[PubMed PMID: 32293641]
[23]
Boudreaux BW, Pincelli TP, Bhullar PK, Patel MH, Brumfiel CM, Li X, Heckman MG, Pittelkow MR, Mangold AR, Sluzevich JC. Secukinumab for the treatment of adult-onset pityriasis rubra pilaris: a single-arm clinical trial with transcriptomic analysis. The British journal of dermatology. 2022 Nov:187(5):650-658. doi: 10.1111/bjd.21708. Epub 2022 Jul 15
[PubMed PMID: 35701384]
[24]
Blauvelt A, Nahass GT, Pardo RJ, Kerdel FA. Pityriasis rubra pilaris and HIV infection. Journal of the American Academy of Dermatology. 1991 May:24(5 Pt 1):703-5
[PubMed PMID: 1869640]
[25]
Misery I, Faure M, Claidy A. Pityriasis rubra pilaris and human immunodeficiency virus infection--type 6 pityriasis rubra pilaris? The British journal of dermatology. 1996 Dec:135(6):1008-9
[PubMed PMID: 8983330]
[26]
Craiglow BG, Boyden LM, Hu R, Virtanen M, Su J, Rodriguez G, McCarthy C, Luna P, Larralde M, Humphrey S, Holland KE, Hogeling M, Hidalgo-Matlock B, Ferrari B, Fernandez-Faith E, Drolet B, Cordoro KM, Bowcock AM, Antaya RJ, Ashack K, Ashack RJ, Lifton RP, Milstone LM, Paller AS, Choate KA. CARD14-associated papulosquamous eruption: A spectrum including features of psoriasis and pityriasis rubra pilaris. Journal of the American Academy of Dermatology. 2018 Sep:79(3):487-494. doi: 10.1016/j.jaad.2018.02.034. Epub 2018 Mar 1
[PubMed PMID: 29477734]
[27]
Velasco RC, Shao C, Greiling TM. Patient-reported cutaneous signs and symptoms of adult pityriasis rubra pilaris and correlation with quality of life and clinician-reported severity: A cross-sectional study. Journal of the American Academy of Dermatology. 2024 Jan:90(1):200-202. doi: 10.1016/j.jaad.2023.08.100. Epub 2023 Sep 23
[PubMed PMID: 37748557]
Level 2 (mid-level) evidence
[28]
Ji-Xu A, Lei DK, Worswick S, Maloney NJ, Kim MM, Cutler L. Patient and disease characteristics associated with psychiatric symptoms and impaired quality of life in pityriasis rubra pilaris. The British journal of dermatology. 2022 Dec:187(6):1024-1026. doi: 10.1111/bjd.21792. Epub 2022 Aug 18
[PubMed PMID: 35895853]
Level 2 (mid-level) evidence
[29]
Griffiths WA. Pityriasis rubra pilaris--an historical approach. 2. Clinical features. Clinical and experimental dermatology. 1976 Mar:1(1):37-50
[PubMed PMID: 1269179]
[30]
Batra J, Gulati S, Sarangal R, Chopra D, Puri S, Kaur R. Utility of Dermoscopy in the Diagnosis of Erythroderma: A Cross-Sectional Study. Indian dermatology online journal. 2023 Nov-Dec:14(6):821-828. doi: 10.4103/idoj.idoj_678_22. Epub 2023 Oct 17
[PubMed PMID: 38099018]
Level 2 (mid-level) evidence
[31]
Gleeson CM, Chan I, Griffiths WA, Bunker CB. Eruptive seborrhoeic keratoses associated with erythrodermic pityriasis rubra pilaris. Journal of the European Academy of Dermatology and Venereology : JEADV. 2009 Feb:23(2):217-8. doi: 10.1111/j.1468-3083.2008.02799.x. Epub 2008 May 13
[PubMed PMID: 18482315]
[32]
Sahin MT, Oztürkcan S, Ermertcan AT, Saçar T, Türkdogan P. Transient eruptive seborrhoeic keratoses associated with erythrodermic pityriasis rubra pilaris. Clinical and experimental dermatology. 2004 Sep:29(5):554-5
[PubMed PMID: 15347352]
[33]
Richey PM, Fairley JA, Stone MS. Transformation from pityriasis rubra pilaris to erythema gyratum repens-like eruption without associated malignancy: A report of 2 cases. JAAD case reports. 2018 Oct:4(9):944-946. doi: 10.1016/j.jdcr.2018.07.009. Epub 2018 Oct 10
[PubMed PMID: 30345340]
Level 3 (low-level) evidence
[34]
Davis AE, Raine BE, Swartzman I, Bogner PN, Nazareth M. Rethinking pityriasis rubra pilaris as a paraneoplastic syndrome: Two cases of pityriasis rubra pilaris with concomitant underlying malignancy. JAAD case reports. 2023 Feb:32():90-95. doi: 10.1016/j.jdcr.2022.12.007. Epub 2022 Dec 27
[PubMed PMID: 36691586]
Level 3 (low-level) evidence
[35]
Gelmetti C, Schiuma AA, Cerri D, Gianotti F. Pityriasis rubra pilaris in childhood: a long-term study of 29 cases. Pediatric dermatology. 1986 Dec:3(6):446-51
[PubMed PMID: 3562357]
Level 3 (low-level) evidence
[36]
Piamphongsant T, Akaraphant R. Pityriasis rubra pilaris: a new proposed classification. Clinical and experimental dermatology. 1994 Mar:19(2):134-8
[PubMed PMID: 8050142]
[37]
Allison DS, El-Azhary RA, Calobrisi SD, Dicken CH. Pityriasis rubra pilaris in children. Journal of the American Academy of Dermatology. 2002 Sep:47(3):386-9
[PubMed PMID: 12196748]
[38]
Gelmetti C, Cerri D. Pityriasis rubra pilaris: the problem of its classification. Journal of the American Academy of Dermatology. 1990 Dec:23(6 Pt 1):1186-8
[PubMed PMID: 2091649]
[39]
Sánchez-Regaña M, Fuentes CG, Creus L, Salleras M, Umbert P. Pityriasis rubra pilaris and HIV infection: a part of the spectrum of HIV-associated follicular syndrome. The British journal of dermatology. 1995 Nov:133(5):818-9
[PubMed PMID: 8555047]
[40]
Shao C, Velasco R, Greiling TM. The individual Pityriasis Rubra Pilaris area and severity index (iPRPASI): validity, reliability, and responsiveness of a novel patient-reported severity tool. Archives of dermatological research. 2023 Dec:315(10):2933-2935. doi: 10.1007/s00403-023-02682-7. Epub 2023 Aug 2
[PubMed PMID: 37532946]
[41]
Goldsmith LA, Weinrich AE, Shupack J. Pityriasis rubra pilaris response to 13-cis-retinoic acid (isotretinoin). Journal of the American Academy of Dermatology. 1982 Apr:6(4 Pt 2 Suppl):710-5
[PubMed PMID: 7040513]
[42]
Kromer C, Sabat R, Celis D, Mössner R. Systemic therapies of pityriasis rubra pilaris: a systematic review. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2019 Mar:17(3):243-259. doi: 10.1111/ddg.13718. Epub 2018 Dec 6
[PubMed PMID: 30520557]
Level 1 (high-level) evidence
[43]
Engelmann C, Elsner P, Miguel D. Treatment of pityriasis rubra pilaris type I: a systematic review. European journal of dermatology : EJD. 2019 Oct 1:29(5):524-537. doi: 10.1684/ejd.2019.3641. Epub
[PubMed PMID: 31789274]
Level 1 (high-level) evidence
[44]
De Rosa A, Gambardella A, Licata G, Alfano R, Argenziano G. Successful treatment of Pityriasis rubra pilaris with brodalumab. The Australasian journal of dermatology. 2020 May:61(2):e249-e251. doi: 10.1111/ajd.13215. Epub 2019 Dec 18
[PubMed PMID: 31851372]
[45]
Saad M, Spurr A, Lipson J. Pityriasis rubra pilaris partially responsive to treatment with upadacitinib: A case report. SAGE open medical case reports. 2023:11():2050313X231160927. doi: 10.1177/2050313X231160927. Epub 2023 Mar 29
[PubMed PMID: 37009550]
Level 3 (low-level) evidence
[46]
Kromer C, Schön MP, Mössner R. Bimekizumab in refractory pityriasis rubra pilaris. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2024 Jan:22(1):102-104. doi: 10.1111/ddg.15252. Epub 2023 Dec 8
[PubMed PMID: 38066410]
[47]
Pilz AC, Seiringer P, Biedermann T, Eyerich K. Treatment of Pityriasis Rubra Pilaris With Guselkumab. JAMA dermatology. 2019 Dec 1:155(12):1424-1426. doi: 10.1001/jamadermatol.2019.2774. Epub
[PubMed PMID: 31577328]
[48]
Ricar J, Cetkovska P. Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab. The British journal of dermatology. 2021 May:184(5):e148. doi: 10.1111/bjd.19681. Epub 2020 Dec 4
[PubMed PMID: 33274752]
[49]
Licata G, Gambardella A, Calabrese G, Pagliuca F, Alfano R, Argenziano G. Refractory Type I pityriasis rubra pilaris treated with tildrakizumab. Clinical and experimental dermatology. 2021 Dec:46(8):1594-1595. doi: 10.1111/ced.14790. Epub 2021 Jul 14
[PubMed PMID: 34101231]
[50]
Zhang YH, Zhou Y, Ball N, Su MW, Xu JH, Zheng ZZ. Type I pityriasis rubra pilaris: upregulation of tumor necrosis factor alpha and response to adalimumab therapy. Journal of cutaneous medicine and surgery. 2010 Jul-Aug:14(4):185-8
[PubMed PMID: 20642989]
[51]
Garcovich S, Di Giampetruzzi AR, Antonelli G, Garcovich A, Didona B. Treatment of refractory adult-onset pityriasis rubra pilaris with TNF-alpha antagonists: a case series. Journal of the European Academy of Dermatology and Venereology : JEADV. 2010 Aug:24(8):881-4. doi: 10.1111/j.1468-3083.2009.03511.x. Epub 2009 Nov 30
[PubMed PMID: 20002243]
Level 2 (mid-level) evidence
[52]
Krase IZ, Cavanaugh K, Curiel-Lewandrowski C. Treatment of Refractory Pityriasis Rubra Pilaris With Novel Phosphodiesterase 4 (PDE4) Inhibitor Apremilast. JAMA dermatology. 2016 Mar:152(3):348-50. doi: 10.1001/jamadermatol.2015.3405. Epub
[PubMed PMID: 26536384]
[53]
Ying Y, Yu-Hua L, Xiao-Yan W, Su-Chun H. A case of pityriasis rubra pilaris treated with tofacitinib after failure with acitretin and ixekizumab. The Australasian journal of dermatology. 2023 Aug:64(3):445-447. doi: 10.1111/ajd.14076. Epub 2023 May 18
[PubMed PMID: 37200390]
Level 3 (low-level) evidence
[54]
Magro C, Cheng E, Kline MA, Belsito DV, Goldman B, Varghese M, Crowson AN, Momtahen S. Follicular psoriasis: a report of 5 cases and review of the literature, likely an under-recognized yet distinctive variant of psoriasis. Dermatology online journal. 2020 Jul 15:26(7):. pii: 13030/qt5103211m. Epub 2020 Jul 15
[PubMed PMID: 32898397]
Level 3 (low-level) evidence
[55]
Halper K, Wright B, Maloney NJ, Kim MM, Ravi V, Worswick S, Lei DK. Characterizing Disease Features and Other Medical Diagnoses in Patients With Pityriasis Rubra Pilaris. JAMA dermatology. 2020 Dec 1:156(12):1373-1374. doi: 10.1001/jamadermatol.2020.3426. Epub
[PubMed PMID: 32965475]
[56]
Conaghan PG, Sommer S, McGonagle D, Veale D, Waldmann H, Hale G, Goodfield M, Emery P, Isaacs J. The relationship between pityriasis rubra pilaris and inflammatory arthritis: case report and response of the arthritis to anti-tumor necrosis factor immunotherapy. Arthritis and rheumatism. 1999 Sep:42(9):1998-2001
[PubMed PMID: 10513817]
Level 3 (low-level) evidence
[57]
Haro R, Revelles JM, Fariña Mdel C, Martín L, Requena L. Wong's dermatomyositis: a new case and review of the literature. International journal of dermatology. 2013 Apr:52(4):466-70. doi: 10.1111/j.1365-4632.2011.05244.x. Epub 2012 Dec 11
[PubMed PMID: 23230970]
Level 3 (low-level) evidence
[58]
Ross NA, Chung HJ, Li Q, Andrews JP, Keller MS, Uitto J. Epidemiologic, Clinicopathologic, Diagnostic, and Management Challenges of Pityriasis Rubra Pilaris: A Case Series of 100 Patients. JAMA dermatology. 2016 Jun 1:152(6):670-5. doi: 10.1001/jamadermatol.2016.0091. Epub
[PubMed PMID: 26963004]
Level 2 (mid-level) evidence
[59]
Yang CC, Shih IH, Lin WL, Yu YS, Chiu HC, Huang PH, Cheng YW, Lee JY, Chen W. Juvenile pityriasis rubra pilaris: report of 28 cases in Taiwan. Journal of the American Academy of Dermatology. 2008 Dec:59(6):943-8. doi: 10.1016/j.jaad.2008.07.054. Epub 2008 Sep 25
[PubMed PMID: 18819727]
Level 3 (low-level) evidence