Continuing Education Activity
Creutzfeldt-Jakob disease (CJD) is a rare, fatal degenerative brain disorder caused by prion proteins. This condition belongs to a group of transmissible spongiform encephalopathies affecting people worldwide, with an incidence of 1 case per million per year. Approximately 350 cases are diagnosed annually in the United States. Death occurs in nearly 70% of patients within a year. Prompt and accurate diagnosis of CJD impacts intervention strategies and overall outcomes.
This activity for healthcare professionals is designed to enhance learners' competence in recognizing and managing CJD. Valuable insights will be gained that will equip learners to work as part of an interprofessional team caring for patients with this condition.
Objectives:
Differentiate between the sporadic, genetic, and infectious forms of Creutzfeldt-Jakob disease in terms of etiology and presentation.
Identify the role of prion proteins in the pathophysiology of Creutzfeldt-Jakob disease.
Describe the Creutzfeldt-Jakob disease evaluation process from the clinical encounter to the interpretation of laboratory and imaging results.
Collaborate with an interprofessional team in treating patients affected by Creutzfeldt-Jakob disease.
Introduction
Creutzfeldt-Jakob disease (CJD) is a rapidly progressive, rare, transmissible, and universally fatal neurodegenerative condition caused by prion proteins. The condition has a long incubation period.[1][2] CJD was first described in 1920 by Hans Creutzfeldt and later in 1921 and 1923 by Alfons Jakob. Later, Clearance J. Gibbs started using the term "Creutzfeldt-Jacob disease" because the acronym "CJD" was closer to his initials.[3][4]
CJD primarily affects the central nervous system (CNS). The CNS' chief functional unit is the neuron, a unique cell type that can receive, store, and transmit information. CNS neurons do not regenerate, although some regions of the brain can heal to a limited extent due to the presence of stem cells. Attributes that make the CNS unique from other organ systems include the following:
- Cerebral blood flow autoregulation
- Having the cranium as bony protection
- Special metabolic substrate requirements
- Absence of a true lymphatic system
- Cerebrospinal fluid (CSF) circulation
- Little immunologic surveillance
- Distinct injury response and tissue repair mechanisms
Neurons in the brain are topographically organized, with functional domains existing in anatomically defined regions. For example, the cerebral cortex controls voluntary movements, while the hypothalamus plays a huge role in autonomic responses. The brain cells' somatotopic organization allows various body areas to receive sensory and motor input from the CNS, which is valuable in localizing neurologic lesions.
Neurons differ in structure and size. Synapses are formed by axons and dendrites, which greatly differ in number from cell to cell. Nissl bodies, which also vary in number, are vital to nerve cell protein and neurotransmitter synthesis. Neurofilaments maintain the cytoskeleton and play a role in nerve conduction.
Glia are cells that support the neurons. They include the following:
- Astrocytes: star-shaped glial cells that supply nutrients to the neurons, act as nerve detoxifiers and electrical insulators, protect from harmful macromolecules, and contribute to CNS repair and scar formation.
- Oligodendrocytes: myelinate the CNS axons.
- Ependymal cells: line the ventricles and control CSF production and flow.
- Microglia: comprise the CNS' macrophage system.
CJD neuronal inclusions damage brain neurons, manifesting with nonspecific prodromal symptoms early in the disease course and neurologic changes, particularly myoclonus, in the advanced stages.
Etiology
A prion protein (PrP) is a normal neuron protein with a predominantly α-helical and random coil composition. Proteinaceous infectious particles, also called "prions," are self-propagating proteins lacking nucleic acid and are mostly comprised of proteinase K-resistant β-pleated sheet aggregates. Prions reproduce by associating with normal PrP cellular isoforms, converting α-helices into indigestible β-pleated sheets. These particles cause CJD and other transmissible spongiform encephalopathies like bovine spongiform encephalopathy (mad cow disease), kuru, and scrapie.
CJD may be classified based on the mode of transmission. Sporadic CJD, the most common type (∼85%), is due to misfolding of normal PrP isoforms with no apparent triggers. Subtypes of sporadic CJD include sporadic fatal insomnia and variably protease-sensitive prionopathy. Genetic CJD, the second most common type (∼10-15%), arises from a heritable genetic mutation. Subtypes of this condition include familial CJD, fatal familial insomnia, and Gerstmann-Sträussler-Scheinker syndrome.
Infectious CJD accounts for less than 1% of cases, and it arises from prion transmission by an external source. Subtypes of infectious CJD include kuru, iatrogenic CJD, and variant CJD. Kuru is a disease of Papua New Guinea's Fore people, who consumed the brains of dead relatives as part of ritualistic cannibalism before the practice was banned in the 1950s. Iatrogenic CJD arises from inadvertent prion inoculation during procedures. Variant CJD is associated with the ingestion of infected beef, a mechanism similar to bovine spongiform encephalopathy. Most documented variant CJD cases are from the United Kingdom and France.[5][6][7]
Epidemiology
CJD affects about 1 individual per million per year worldwide. Approximately 350 cases are diagnosed annually in the United States. Sporadic CJD is the most common form of human prion disease. The condition has a mean onset age of 62, although it has also been reported in younger and older age groups.[8][9] Sporadic CJD has a 1:1 male-to-female ratio. Approximately 1 to 2 new sporadic CJD cases appear per 1,000,000 individuals worldwide annually.[10] Death occurs in nearly 70% of patients within a year of onset. The mean survival of sporadic CJD is 4 to 8 months, with 90% of patients dying within a year.
Genetic CJD is the second most common type of this condition. Patients often have a family history and autosomal-dominant PRNP gene mutations. Acquired cases are seen in less than 1% of cases, usually in young adults with a mean age of 29.
Pathophysiology
Normal cellular prion protein (PrPc) transforms into the disease-causing form PrP scrapie (PrPSc) either spontaneously or as a result of PrPSc infection. PrPSc self-propagates and accumulates throughout the brain. The highly chemically stable β-pleated aggregates cause derangements in intracellular protein folding, ubiquitination, and trafficking in affected neurons. Additionally, astrocytes may swell and degrade in reaction to prion-induced injury. Neurodegeneration results from these changes.[11]
Histopathology
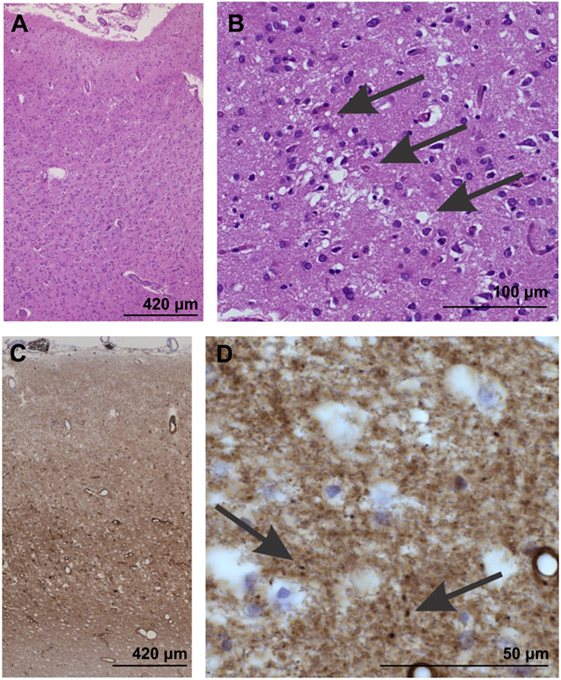
Gross examination of affected brains may not reveal abnormalities. However, the following features are commonly observed during light microscopy (see Image. Neuropathological Confirmation of Creutzfeldt-Jacob Disease):
- Vacuolation or spongiform degeneration, most notably in the cerebral cortex, caudate nucleus, thalamus, putamen, and cerebellum's molecular layer
- Neuronal loss
- Astrocytic gliosis or fibrous proliferation of the astrocytes, mostly in the gray matter
Amyloid plaques are found in some individuals.
History and Physical
CJD presents variably, depending on the subtype. The manifestations arise from the involvement of various parts of the CNS. The table below summarizes the symptoms that may manifest in patients with CJD.
Table. Symptoms of Creutzfeldt-Jakob Disease
Neuropsychiatric symptoms:
- Dementia
- Behavioral abnormalities
- Aphasia
- Apraxia
- Frontal lobe syndromes
- Impaired concentration, memory, and judgment
- Depression
- Apathy
- Anxiety
- Sleep disturbances
- Psychotic features, especially visual hallucinations [12][13][14][15]
|
|
Involuntary movements:
- Myoclonus
- Tremor
- Choreoathetosis
- Hemiballismus [31]
|
| Cerebellar manifestations:[16]
|
Signs related to the corticospinal tract:
- Extensor plantar responses
- Dystonia
- Rigidity
- Hypokinesia
|
Atypical features include cranial nerve abnormalities, peripheral nervous system involvement, and vestibulocochlear dysfunction. Although found rarely in association with CJD, these manifestations should raise suspicion of another diagnosis. The neurologic examination must be as detailed as possible for all patients suspected of CJD.
Sporadic CJD
In the early stages of sporadic CJD, patients may develop nonspecific symptoms like vertigo, headache, fatigue, and sleep disorders. However, memory problems, agitation, irritability, depression, apathy, mood swings, and sensory changes like vision loss can also occur. As the disease advances, patients may develop rapidly worsening confusion, disorientation, and cognitive problems. Most patients manifest with coordination and movement abnormalities, including ataxia, involuntary jerky movements, myoclonus, muscle stiffness, and involuntary muscle twitching. Myoclonus persists during sleep and may be elicited by loud sounds or bright lights.
Extrapyramidal symptoms like bradykinesia, dystonia, and rigidity may occur. Patients gradually lose mobility and speech and progress into a comatose state. Certain infections, such as pneumonia, can lead to death.
Patients with sporadic CJD are typically between 55 and 75 years old. Death occurs within a year of onset, with the median duration of illness being 4 to 5 months. The median age of death in individuals with sporadic CJD is 68 years. Sporadic CJD is similar to dementia in presentation but progresses much more rapidly.
CJD subtyping is based on genetic polymorphism, prion protein characteristics, and associated symptoms. Methionine/methionine type 1 (MM1) and methionine/valine type 1 (MV1) comprise 70% of cases and correlate with the classic CJD phenotype. The condition has a mid- to late-life onset and presents with rapidly progressive dementia (RPD) and early myoclonus and ataxia.
Methionine/valine type 2 (MV2) is the kuru plaque variant, comprising 10% of sporadic CJD cases. The condition presents with progressive dementia with prominent psychiatric features and has a longer duration (about 17 months).[17] The valine/valine type 2 (VV2) variant is also known as the ataxic variant. The condition presents with ataxia at the onset or as an isolated feature. The VV2 subtype has a duration of illness of about 7 to 9 months.[18]
The methionine/methionine type 2 (MM2) subtype may be classified as either thalamic or cortical. The thalamic MM2 subtype is also known as sporadic fatal insomnia and is present in 2% of cases. The mean disease duration is 15.6 months. The most frequent symptoms include psychomotor hyperactivity, ataxia, insomnia, and cognitive impairment, resembling fatal familial insomnia.[19] The cortical MM2 subtype also comprises 2% of sporadic CJD cases. Dementia is the predominant manifestation of this condition. The average disease duration is 15.7 months. Visual and cerebellar signs are rarely described at presentation.[20]
The valine/valine 1 (VV1) subtype comprises 1% of cases and has a younger age of onset. Individuals with this condition present with progressive dementia. The VV1 subtype has an average duration of 15.3 months.[21]
Inherited CJD
Genetic CJD has phenotypic variability that may be attributed to the low penetrance of PRNP mutations. Patients with genetic CJD are usually younger than individuals with sporadic CJD, manifesting behavioral and cognitive changes initially, and incoordination and movement abnormalities over the next few months. A family history of similar neurologic manifestations may be elicited.
Inherited CJD is fatal, though the duration of illness varies individually. For example, Gerstmann–Straussler–Scheinker syndrome has a slow progression, and death may be delayed for up to 10 years.[32]
Variant CJD
Patients with variant CJD are often younger than patients with sporadic CJD, initially presenting with psychiatric symptoms, behavioral changes, and painful dysesthesias. Movement disorders may develop early, but dementia is usually a late sign. A history of a neurosurgical procedure or ingestion of infected meat may be elicited. The median duration of illness is between 13 and 14 months, while the median age at death is 28 years.
Evaluation
Creutzfeldt-Jakob disease is often a diagnostic challenge as it presents similarly to other conditions presenting with RPD. The following are the recommended initial screening tests for evaluating RPD:
- Complete blood count
- Complete metabolic panel
- Blood magnesium
- Rapid plasma reagin
- Erythrocyte sedimentation rate
- Antinuclear antibody
- C-reactive protein
- Thyroid function tests
- Vitamin B12 level
- HIV test
- Lyme disease titer
- Autoimmune antibodies
- Urinalysis
- CSF studies, including glucose, oligoclonal bands, and cell count with differential
- CSF 14-3-3 protein (a test for prion disease)
- Venereal disease research laboratory (VDRL) test
Among imaging studies, computed tomography (CT) may be ordered initially. However, brain magnetic resonance imaging (MRI) with or without contrast and the related modalities fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted imaging (DWI) can give better detail about the affected regions. CJD may be diagnosed when imaging is combined with clinical, laboratory, and electroencephalogram (EEG) findings.[22][23][24]
The World Health Organization published diagnostic criteria for CJD in 1998, which relied on the clinical, EEG, and CSF findings. However, advanced diagnostic procedures like MRI and genetic testing have made those criteria obsolete.
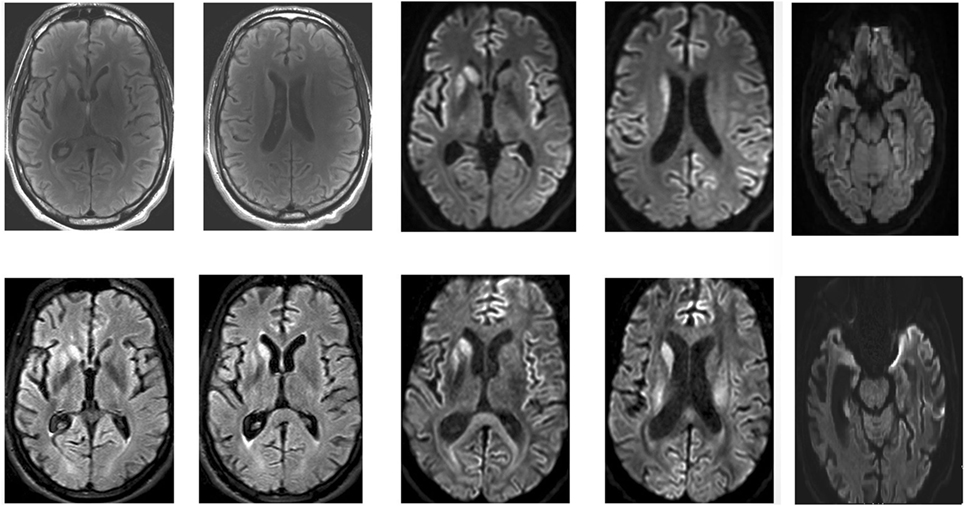
Brain MRI is a more sensitive and specific test for variant CJD than CSF 14–3–3 protein and was found to be accurate in about 90% of cases (see Image. Brain MRI of a Patient with Early-Stage Creutzfeldt-Jakob Disease).[25] Brain MRI with T2-weighted scanning, DWI, and apparent diffusion coefficient (ADC) sequences often reveal abnormalities in the cortical gray matter (cortical ribboning) and deep nuclei in sporadic CJD. MRI with DWI or FLAIR imaging has a sensitivity of 98% and specificity of 93%. DWI typically demonstrates hyperintensities within the basal ganglia, thalamus, and cortex. The "hockey stick" or "pulvinar" sign indicates variant, infectious, or acquired CJD, though it is also seen in other forms of CJD. CSF 14-3-3 can be more sensitive for CJD than other prion diseases when combined with the typical EEG findings.
CSF protein biomarkers, including the 14–3–3 protein, total tau (T-tau), and neuron-specific enolase (NSE), are markers of rapid neurodegeneration. These tests can help diagnose CJD but are not specific to the condition. An elevated tau level (greater than 1150 picogram/mL) has more accuracy and specificity than 14-3-3 protein as a diagnostic test for CJD, although both tests can produce significant false-negative and false-positive results.[26]
In 2012, the American Academy of Neurology recommended ordering CSF 14-3-3 only when CJD is strongly suspected. A recent comparison of these non-prion-specific CSF biomarkers with MRI found that DWI had a diagnostic accuracy of 97%, higher than the T-tau (79.6%), 14-3-3 protein (70.4%), and NSE (71.4%) tests. Detection of these traditional surrogate marker proteins is accurate in approximately three-fourths of cases. Routine CSF analysis that includes glucose, total protein, white blood cell count, total cell count, and oligoclonal IgG are generally unremarkable in CJD patients.
The National Prion Disease Pathology Surveillance Center launched a new diagnostic test in April 2015 called "second-generation Real Time-Quaking-Induced Conversion (RT-QuIC)," which is highly sensitive and specific for CJD. RT-QuIC can accurately detect pathogenic prion protein in the CSF of patients with CJD. RT-QuIC directly detects the pathogenic prion protein, in contrast to the 14-3-3 protein, T-tau, and NSE, which are indirect tests.
A few studies reveal that RT-QuIC has modest sensitivity (greater than 80%) but high specificity (approximately 98%) for sporadic CJD. Most patients with the VV2 sporadic CJD subtype have negative CSF RT-QuIC tests. Although it is not as sensitive as the MRI, RT-QuIC is often positive in many forms of genetic prion disease, some of which lack the classic sporadic CJD MRI findings.
RT-QuIC might be more sensitive using olfactory epithelium brushings than the CSF as a specimen. Recent studies have shown that RT-QuIC is comparable to brain biopsy in terms of accuracy in diagnosing CJD. RT-QulC is less invasive compared to brain biopsy. However, the existing CJD guidelines do not include newer, less invasive diagnostic modalities.
EEG is not as sensitive as brain MRI or CSF studies in detecting CJD, though the typical findings associated with the condition in the late stages are periodic sharp wave complexes (PSWCs). Bi- or triphasic PSWC is found in 67% to 95% of patients with sporadic CJD. This finding is considered supportive rather than diagnostic and is characterized by the following features:
- Generalized or lateralized complexes
- A minimum of 5 repetitive intervals with a duration difference of less than 500 milliseconds to exclude semiperiodic activity
- Strictly periodic cerebral potentials, the majority with an intercomplex interval of 500 to 2000 milliseconds and a duration of 100 to 600 milliseconds
Among patients with sporadic CJD, the MM1 and MV2 subtypes frequently manifest with PSWCs, while the MV2 subtype exhibits PSWCs infrequently. PSWCs are not found in patients with Gerstmann-Sträussler-Scheinker syndrome, variant CJD, kuru, or fatal familial insomnia. PSWCs are observed occasionally in patients with genetic CJD, particularly in patients with the codon 200 mutation.[27]
Brain tissue biopsy or postmortem examination of the brain confirms the diagnosis of CJD. However, not all areas of the brain are affected by the disease. Imaging studies must target subcortical structures where abnormal features are most likely found. Surgery can be risky and may not always obtain the affected brain tissue. CJD confirmation does not change the patient's clinical outcome. Thus, a brain biopsy is only indicated when a reversible condition is suspected in the differential.
Prions have been detected in the blood and urine of patients with symptomatic variant CJD. Caution should be exercised when handling body fluids and tissues from patients with variant CJD until prion infectivity has been ruled out.
Treatment / Management
CJD has no definitive treatment, and supportive care is the mainstay of management. Most trial drugs for CJD have not demonstrated any clear benefit to date. However, intraventricular pentosan polysulfate has been shown in rodent studies to inihibit PrPSc formation. An apparent survival extension of 37 to 114 months was observed in four patients.[28] More research is needed to find the cure for this fatal condition.
Early detection of a PRNP mutation may help families at risk for the genetic form of CJD. Patients affected by the condition can make earlier arrangements for end-of-life concerns. Psychosocial support and supportive care may improve patients' quality of life. Genetic counseling and family planning help prevent disease transmission to the offspring of individuals with PRNP mutation.
Differential Diagnosis
RPD has a broad differential, including vascular, neurodegenerative, autoimmune, infectious, thromboembolic, neoplastic, iatrogenic, and toxic metabolic conditions. Vascular conditions like stroke, multiple infarcts, cerebral myeloid angioplasty, or hypertensive encephalopathy can likewise lead to RPD. Vasculitis and intravascular lymphoma may also manifest as RPD.
These conditions may be distinguished from CJD by a thorough medical evaluation. Imaging studies can help rule out vascular and neoplastic causes. Blood and CSF examinations can detect autoimmune, infectious, metabolic, and neurodegenerative conditions.[29]
Prognosis
CJD's prognosis is extremely poor despite all the advances that have helped understand this disease. The condition is invariably fatal. Death occurs within one year of symptom onset, except in some cases.
Complications
CJD's complications encompass both physical and psychosocial difficulties. Individuals affected by CJD often withdraw from friends and family and ultimately lose their ability to recognize or relate to them. Patients also lose their capacity for self-care and often eventually slip into a coma. CJD has a 100% fatality rate.
Deterrence and Patient Education
CJD is a progressive, fatal disease. Fortunately, very few cases are reported in the USA, so the risk of contracting the disease is extremely low. Strategies revolve around prevention, with blood centers not permitting first-degree relatives of people with CJD from donating blood. Hunters who would consume or handle elk or deer meat should consider having the meat tested before eating it. Families at risk for the genetic type of CJD may practice contraception to prevent disease transmission to their offspring.
Pearls and Other Issues
The most important points to remember when managing CJD are the following:[30]
- CJD is an invariably fatal prion disease.
- The condition has vast presentations, but cases are generally classified as either sporadic, genetic, or infectious.
- CJD's manifestations are primarily neurologic, with dementia, myoclonus, and EEG PSWCs as the most common features.
- Supportive treatment and psychosocial care are the cornerstones of CJD management.
- Early detection does not alter the disease course, though it can improve patients' quality of life and prevent disease transmission in families with the genetic subtype.
- Diagnosis is possible if clinical evaluation is combined with blood and CSF tests, imaging, and EEG.
Prion diseases like CJD should be considered when evaluating patients with RPD.
Enhancing Healthcare Team Outcomes
Treating CJD requires a multidisciplinary approach due to the condition's complex nature and impact on various aspects of a patient's health. The interprofessional team may include the following members:
- Neurologists: diagnose and manage the condition and counsel patients and families about how CJD affects brain function
- Neurosurgeons: may perform neurosurgical procedures for symptom management in patients with CJD
- Infectious disease specialists: may contribute their expertise in managing prion transmission and implementing infection control measures
- Intensivists: provide treatment to CJD patients in intensive care
- Mental health practitioners: provide emotional and psychological support to patients with CJD and their families, helping them cope with the mental health challenges associated with the disease
- Neuroradiologists: lend their expertise in interpreting imaging studies, aiding in CJD diagnosis and monitoring of brain changes
- Pathologists: examine brain tissue samples to confirm the diagnosis of CJD
- Nurses: provide direct patient care, administer treatments, monitor symptoms, and support patients and families. Nurses often serve as a crucial link between the healthcare team, patients, and their families.
- Rehabilitation team: physical and occupational therapists help manage symptoms and maintain functional abilities
- Speech-language pathologists: assist in managing communication difficulties that can arise due to CJD-related neurological changes
- Palliative care specialists: provide symptom management, pain relief, and support to patients with CJD and families facing end-of-life care. These providers focus on improving the quality of life for patients with CJD.
- Social workers: assist the team in coordinating care. These professionals also aid patients and their families in accessing care and navigating the challenges associated with CJD.
- Genetic counselors: provide information, support, and counseling about familial risks and genetic testing to patients at risk for the inherited type of CJD
Collaboration among team members is vital in rendering the best care to individuals affected by CJD.
Most patients with CJD die within a year from the onset of symptoms. Preparations for hospice care should be addressed immediately. Counseling for the family is essential because the condition has no effective cure, and death is inevitable. Interprofessional collaboration can help smooth the fatal course of this disease with family counseling and appropriate palliative care.