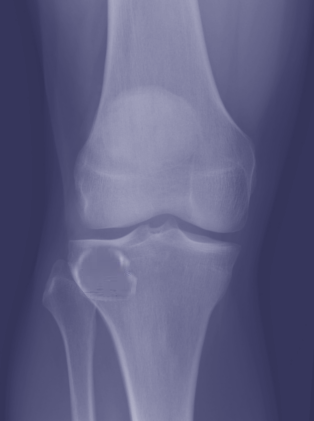
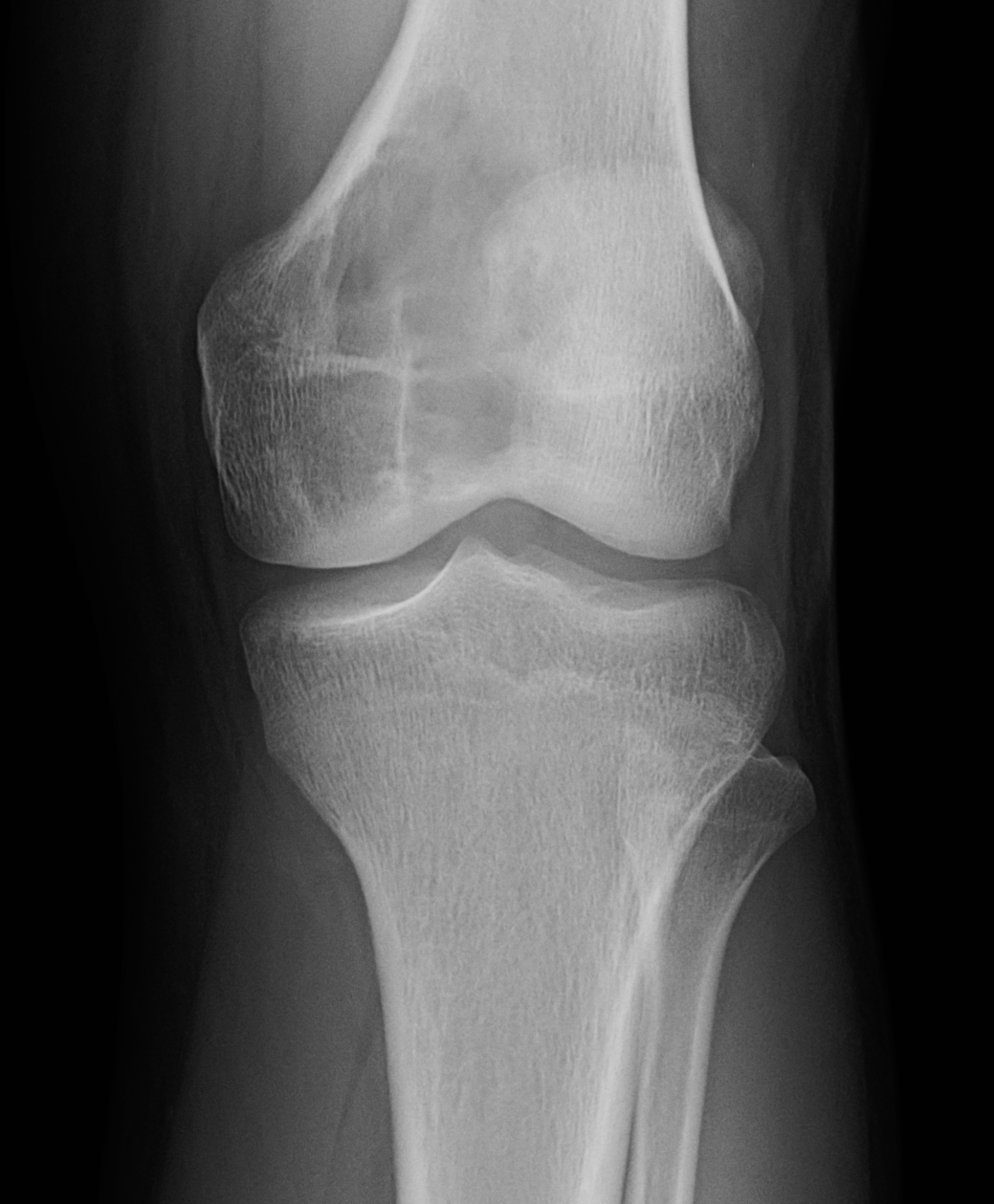
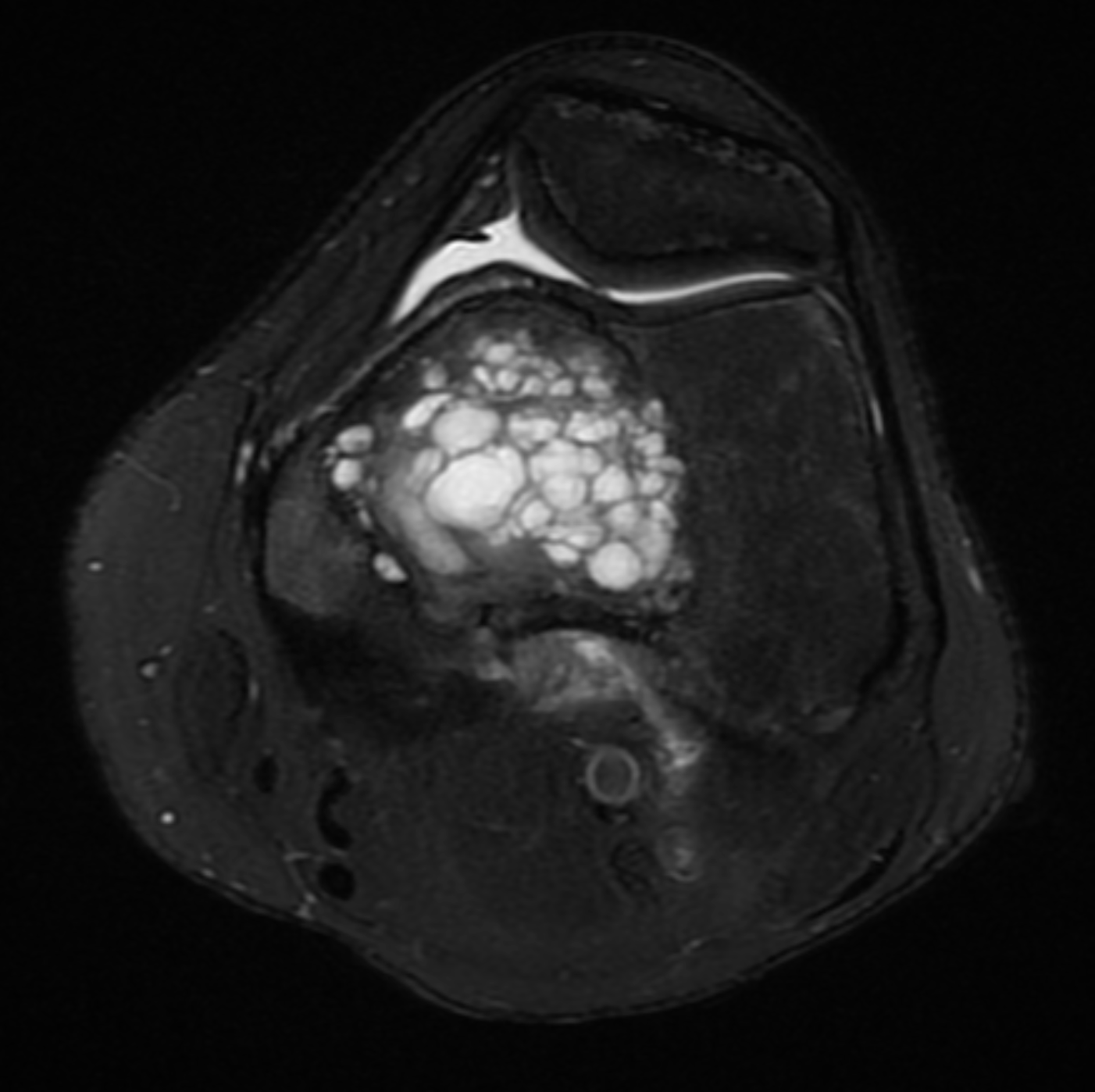
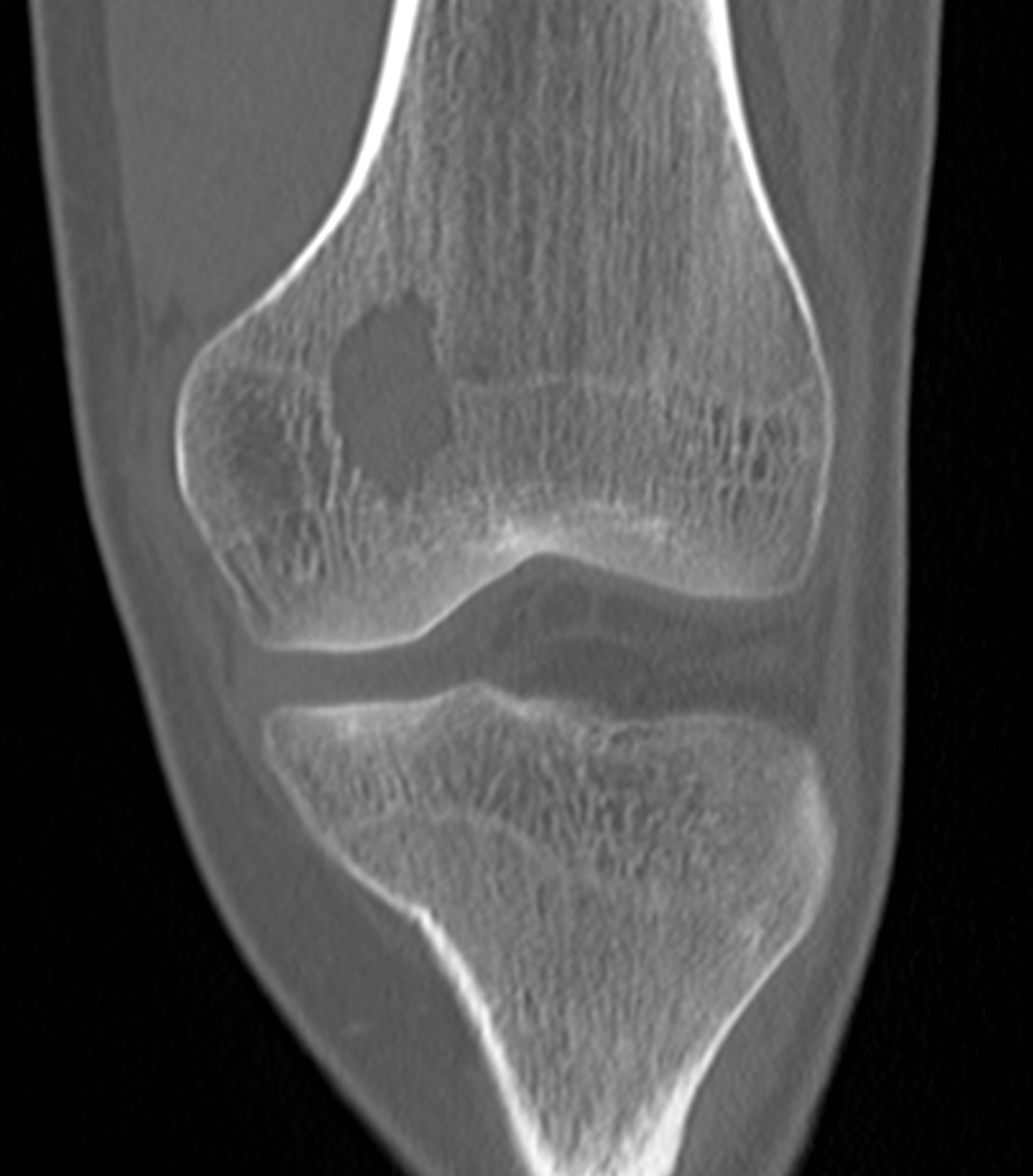
[1]
Chen W, DiFrancesco LM. Chondroblastoma: An Update. Archives of pathology & laboratory medicine. 2017 Jun:141(6):867-871. doi: 10.5858/arpa.2016-0281-RS. Epub
[PubMed PMID: 28557595]
[2]
Jaffe HL, Lichtenstein L. Benign Chondroblastoma of Bone: A Reinterpretation of the So-Called Calcifying or Chondromatous Giant Cell Tumor. The American journal of pathology. 1942 Nov:18(6):969-91
[PubMed PMID: 19970672]
[3]
Dahlin DC, Ivins JC. Benign chondroblastoma. A study of 125 cases. Cancer. 1972 Aug:30(2):401-13
[PubMed PMID: 5051664]
Level 3 (low-level) evidence
[4]
De Mattos CB, Angsanuntsukh C, Arkader A, Dormans JP. Chondroblastoma and chondromyxoid fibroma. The Journal of the American Academy of Orthopaedic Surgeons. 2013 Apr:21(4):225-33. doi: 10.5435/JAAOS-21-04-225. Epub
[PubMed PMID: 23545728]
[5]
Sjögren H, Orndal C, Tingby O, Meis-Kindblom JM, Kindblom LG, Stenman G. Cytogenetic and spectral karyotype analyses of benign and malignant cartilage tumours. International journal of oncology. 2004 Jun:24(6):1385-91
[PubMed PMID: 15138578]
[6]
Swarts SJ, Neff JR, Johansson SL, Nelson M, Bridge JA. Significance of abnormalities of chromosomes 5 and 8 in chondroblastoma. Clinical orthopaedics and related research. 1998 Apr:(349):189-93
[PubMed PMID: 9584382]
[7]
Ostrowski ML, Johnson ME, Truong LD, Hicks MJ, Smith FE, Spjut HJ. Malignant chondroblastoma presenting as a recurrent pelvic tumor with DNA aneuploidy and p53 mutation as supportive evidence of malignancy. Skeletal radiology. 1999 Nov:28(11):644-50
[PubMed PMID: 10591928]
[8]
Bloem JL, Mulder JD. Chondroblastoma: a clinical and radiological study of 104 cases. Skeletal radiology. 1985:14(1):1-9
[PubMed PMID: 4023729]
Level 3 (low-level) evidence
[9]
Turcotte RE, Kurt AM, Sim FH, Unni KK, McLeod RA. Chondroblastoma. Human pathology. 1993 Sep:24(9):944-9
[PubMed PMID: 8253461]
[10]
Petsas T, Megas P, Papathanassiou Z. Radiofrequency ablation of two femoral head chondroblastomas. European journal of radiology. 2007 Jul:63(1):63-7
[PubMed PMID: 17482405]
[11]
Brien EW, Mirra JM, Ippolito V. Chondroblastoma arising from a nonepiphyseal site. Skeletal radiology. 1995 Apr:24(3):220-2
[PubMed PMID: 7610417]
[12]
el-Naggar AK, Hurr K, Tu ZN, Teague K, Raymond KA, Ayala AG, Murray J. DNA and RNA content analysis by flow cytometry in the pathobiologic assessment of bone tumors. Cytometry. 1995 Mar 1:19(3):256-62
[PubMed PMID: 7736870]
[13]
Tarkkanen M, Nordling S, Böhling T, Kivioja A, Karaharju E Szymanska J, Elomaa I, Knuutila S. Comparison of cytogenetics, interphase cytogenetics, and DNA flow cytometry in bone tumors. Cytometry. 1996 Sep 15:26(3):185-91
[PubMed PMID: 8889389]
[14]
Mark J, Wedell B, Dahlenfors R, Grepp C, Burian P. Human benign chondroblastoma with a pseudodiploid stemline characterized by a complex and balanced translocation. Cancer genetics and cytogenetics. 1992 Jan:58(1):14-7
[PubMed PMID: 1728944]
[15]
Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, Nik-Zainal S, Martin S, McLaren S, Goodie V, Robinson B, Butler A, Teague JW, Halai D, Khatri B, Myklebost O, Baumhoer D, Jundt G, Hamoudi R, Tirabosco R, Amary MF, Futreal PA, Stratton MR, Campbell PJ, Flanagan AM. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nature genetics. 2013 Dec:45(12):1479-82. doi: 10.1038/ng.2814. Epub 2013 Oct 27
[PubMed PMID: 24162739]
[16]
Amary MF, Berisha F, Mozela R, Gibbons R, Guttridge A, O'Donnell P, Baumhoer D, Tirabosco R, Flanagan AM. The H3F3 K36M mutant antibody is a sensitive and specific marker for the diagnosis of chondroblastoma. Histopathology. 2016 Jul:69(1):121-7. doi: 10.1111/his.12945. Epub 2016 Mar 21
[PubMed PMID: 26844533]
[17]
Konishi E, Nakashima Y, Iwasa Y, Nakao R, Yanagisawa A. Immunohistochemical analysis for Sox9 reveals the cartilaginous character of chondroblastoma and chondromyxoid fibroma of the bone. Human pathology. 2010 Feb:41(2):208-13. doi: 10.1016/j.humpath.2009.07.014. Epub 2009 Oct 3
[PubMed PMID: 19801163]
[18]
Akpalo H, Lange C, Zustin J. Discovered on gastrointestinal stromal tumour 1 (DOG1): a useful immunohistochemical marker for diagnosing chondroblastoma. Histopathology. 2012 Jun:60(7):1099-106. doi: 10.1111/j.1365-2559.2011.04152.x. Epub 2012 Feb 15
[PubMed PMID: 22335248]
[19]
Semmelink HJ, Pruszczynski M, Wiersma-van Tilburg A, Smedts F, Ramaekers FC. Cytokeratin expression in chondroblastomas. Histopathology. 1990 Mar:16(3):257-63
[PubMed PMID: 1692005]
[20]
Clapper AT, DeYoung BR. Chondroblastoma of the femoral diaphysis: report of a rare phenomenon and review of literature. Human pathology. 2007 May:38(5):803-6
[PubMed PMID: 17306329]
[21]
Yang J, Tian W, Zhu X, Wang J. Chondroblastoma in the long bone diaphysis: a report of two cases with literature review. Chinese journal of cancer. 2012 May:31(5):257-64. doi: 10.5732/cjc.011.10402. Epub 2012 Mar 27
[PubMed PMID: 22464651]
Level 3 (low-level) evidence
[23]
Özer D, Arıkan Y, Gür V, Gök C, Akman YE. Chondroblastoma: An evaluation of the recurrences and functional outcomes following treatment. Acta orthopaedica et traumatologica turcica. 2018 Nov:52(6):415-418. doi: 10.1016/j.aott.2018.07.004. Epub 2018 Sep 22
[PubMed PMID: 30249436]
[24]
Angelini A, Arguedas F, Varela A, Ruggieri P. Chondroblastoma of the Foot: 40 Cases From a Single Institution. The Journal of foot and ankle surgery : official publication of the American College of Foot and Ankle Surgeons. 2018 Nov-Dec:57(6):1105-1109. doi: 10.1053/j.jfas.2018.05.005. Epub
[PubMed PMID: 30368424]
Level 3 (low-level) evidence
[25]
Lin PP, Thenappan A, Deavers MT, Lewis VO, Yasko AW. Treatment and prognosis of chondroblastoma. Clinical orthopaedics and related research. 2005 Sep:438():103-9
[PubMed PMID: 16131877]
[26]
Atalar H, Basarir K, Yildiz Y, Erekul S, Saglik Y. Management of chondroblastoma: retrospective review of 28 patients. Journal of orthopaedic science : official journal of the Japanese Orthopaedic Association. 2007 Jul:12(4):334-40
[PubMed PMID: 17657552]
Level 2 (mid-level) evidence
[27]
Hapa O, Karakaşlı A, Demirkıran ND, Akdeniz O, Havitçioğlu H. Operative treatment of chondroblastoma: a study of 11 cases. Acta orthopaedica Belgica. 2016 Mar:82(1):68-71
[PubMed PMID: 26984656]
Level 3 (low-level) evidence
[28]
Riddell RJ, Louis CJ, Bromberger NA. Pulmonary metastases from chondroblastoma of the tibia. Report of a case. The Journal of bone and joint surgery. British volume. 1973 Nov:55(4):848-53
[PubMed PMID: 4766191]
Level 3 (low-level) evidence
[29]
Sailhan F, Chotel F, Parot R, SOFOP. Chondroblastoma of bone in a pediatric population. The Journal of bone and joint surgery. American volume. 2009 Sep:91(9):2159-68. doi: 10.2106/JBJS.H.00657. Epub
[PubMed PMID: 19723993]
[30]
Duttaluri R, Sultanpurkar GP, Raorane H, Vikram H. Malignant chondroblastoma of extraskeletal origin. International journal of applied & basic medical research. 2016 Apr-Jun:6(2):146-8. doi: 10.4103/2229-516X.179028. Epub
[PubMed PMID: 27127749]
[31]
Kyriakos M, Land VJ, Penning HL, Parker SG. Metastatic chondroblastoma. Report of a fatal case with a review of the literature on atypical, aggressive, and malignant chondroblastoma. Cancer. 1985 Apr 15:55(8):1770-89
[PubMed PMID: 3978565]
Level 3 (low-level) evidence