Continuing Education Activity
Ocular melanoma, the second most prevalent form, manifests as a malignant growth of melanocytes in the eye's uveal tract or conjunctiva. Individuals with light-colored irises are at the highest risk, further influenced by factors such as sunlight exposure, genetic predisposition, and certain underlying conditions such as dysplastic nevus syndrome and xeroderma pigmentosum. Detection involves funduscopic examination and imaging modalities, with symptoms ranging from visual impairment to ocular pain.
Although local treatments effectively prevent recurrence, nearly half of patients with uveal melanomas eventually experience metastatic disease. Diagnosis incorporates advanced methods such as ultrasound, optical coherence tomography, funduscopic imaging, and magnetic resonance imaging. Treatment options encompass observation, radiation therapy, and, in severe cases, enucleation with prognosis depending on tumor size, location, and genetic variations. The emergence of targeted therapies presents a promising avenue for managing metastatic ocular melanoma, highlighting the dynamic aspects of its diagnosis and treatment. This activity describes the etiology, epidemiology, presentation, diagnosis, and management of ocular melanoma, equipping healthcare professionals with essential knowledge to mitigate morbidity and mortality in affected patients.
Objectives:
Differentiate ocular melanoma from other ocular conditions based on clinical presentation and diagnostic findings.
Assess the stage and extent of ocular melanoma using standardized staging systems and imaging techniques.
Apply local treatment modalities effectively to prevent recurrence, considering individual patient factors and tumor characteristics.
Collaborate with multidisciplinary team members to ensure comprehensive care for patients with ocular melanoma.
Introduction
Melanoma, marked by the malignant overgrowth of melanocytes, encompasses ocular melanoma, the second most prevalent type after cutaneous melanoma and the most common primary intraocular malignant tumor in adults. Approximately 85% originate in the uveal tract, including the iris, ciliary body, and choroid, with most emerging from the ciliary body or choroid. Melanoma can also arise from the melanocytes in the epithelium of the conjunctival membrane, accounting for less than 10% of ocular melanomas.
The risk is most significant in patients with light-colored irises and is further influenced by factors such as sunlight exposure, genetic predisposition, and underlying illnesses. Ocular melanoma can be diagnosed incidentally through funduscopic examination or when patients present with various visual symptoms such as decreased visual acuity, scotoma, visual field loss, ocular pain, or floaters. Diagnostic tools include ultrasound, optical coherence tomography, and fluorescein angiography. A small percentage of patients require a diagnostic biopsy.
Although localized interventions successfully deter relapse, nearly half of individuals with uveal tumors encounter potential metastatic progression due to early micrometastatic occurrences. Ocular melanoma commonly disseminates hematogenously, with the liver being a primary target. The overall prognosis depends on factors such as tumor size, location, and histologic traits. Importantly, the selection of local treatment does not impact long-term survival outcomes.
Etiology
An increased risk of uveal melanoma is most significantly associated with ocular or oculodermal melanocytosis. Individuals with light-colored eyes, fair skin, and skin that does not tan but burns easily are also at increased risk. The influence of ultraviolet (UV) light exposure remains unclear. If UV light exposure does cause an increased risk, the conferred risk is lower compared to its contribution to cutaneous melanoma. Additional risk factors include cutaneous nevi, cutaneous freckles, iris nevi, and arc welding.[1]
Various genetic risk variants also exist. A recent discovery highlights a genetic correlation between uveal and cutaneous melanoma on chromosome 5p15.33.[2] Researchers believe that 2 potential genes in this area, telomerase reverse transcriptase (TERT) and CLPTM1 regulator of GABA type A receptor forward trafficking (CLPTM1L), may be responsible for conferring an increased risk of ocular melanoma. Recurrent mutations in the TERT promoter are associated with cutaneous melanoma. Other malignancies are associated with variants of CLPTM1L.
Present in 71% to 93% of patients affected by uveal melanoma, variations in the G-protein subunit alpha q (GNAQ) and G-protein subunit alpha 11 (GNA11) genes are the most common genetic variations.[3][4] In addition, variations in BRCA1-associated protein 1 (BAP1), splicing factor 3b subunit 1 (SF3B1), and eukaryotic translation initiation factor 1A X-linked (EIF1AX) are also frequently encountered mutations.
Genetic variations within BAP1, SF3B1, and EIF1AX affect prognosis. Patients with a BAP1 mutation or lacking BAP1 expression have a high risk of developing metastatic disease. An EIF1AX mutation is associated with a low risk of metastatic disease and a better prognosis. Additional indicators of poor prognosis include a gain of chromosome 8q and a loss of chromosome 3 or monosomy 3.[5][6] Other chromosomal variations found in uveal melanoma are chromosome 1p loss and chromosome 6 gain. Patients with an SF3B1 mutation tend to obtain a diagnosis 10 years earlier compared to those with other mutations and are more susceptible to late onset of metastases. Additional but less common genetic variations associated with uveal melanoma include phospholipase C beta 4 (PLCB4), cysteinyl leukotriene receptor 2 (CYSLTR2), RNA-binding motif protein 10 (RBM10), and SRSF2 pseudogene 1 (SRSF2).
Genetic mutations linked to iris melanoma share similarities, with rare occurrences of BAP1 and SF3B1 mutations. An alternative form of the B-Raf proto-oncogene, serine/threonine kinase (BRAF), is connected to iris, conjunctival, and cutaneous melanomas. Conjunctival melanoma involves additional mutations such as the TERT promoter, NRAS proto-oncogene (NRAS), and neurofibromin 1 (NF1). TERT genetic variations significantly impact prognosis. Although some studies reveal an association between BRCA2 pathogenic variants and ocular melanoma, others do not.[7][8].
Epidemiology
Ocular melanoma is the most prevalent primary intraocular malignancy in adults, accounting for 3% to 5% of all melanomas. Uveal melanoma accounts for 85% to 90%, with 5% originating in the iris and the remainder arising from the ciliary body or choroid. The median age of diagnosis is approximately 62, with a peak incidence observed between 70 and 79.
White patients of northern European descent have the highest incidence of disease. In contrast, the incidence among Black patients is low, whereas the incidence in patients who are Asian or Hispanic is considered moderate.[3] Men have a 30% higher incidence compared to females. In the United States, the incidence of ocular melanoma is approximately 5 per 1 million population. Internationally, in countries with large populations of individuals of northern European descent, the incidence is 7.5 per 1 million people annually.
Pathophysiology
Oxidative damage to pigmented tissues, controlled by the degree and type of iris pigmentation, is considered one of the primary mechanisms by which ocular melanoma develops.[2] Primary ocular melanomas arise from melanocytes in the iris, choroid, ciliary body, and conjunctiva. Except for conjunctival melanoma, most evolve from preexisting nevi, but de novo growth is also possible.
Choroidal melanomas typically manifest as dome-shaped tumors, resulting in multilayering of retinal pigment epithelial cells, accumulation of lipofuscin, drusen formation, and detachment of the retinal pigment epithelium. In addition, phagocytic digestion of melanocyte debris induces patches of retinal pigment epithelium color change. These changes ultimately lead to choroidal neovascularization, subretinal exudation, hemorrhage, and fibrous plaque formation. Affected individuals often experience blurred vision, metamorphosis, and visual field loss, primarily due to atrophy and degeneration of the retina. Classically, choroidal melanomas exhibit a mushroom-shaped appearance due to the rupture through Bruch’s membrane and the prolapse of the retinal pigment epithelium into the subretinal space.
Choroidal melanomas tend to exert pressure on the lens, leading to astigmatism, cataracts, and subluxation. In addition, sentinel vessels representing dilated episcleral vessels may be observed in some cases. The majority of conjunctival melanomas arise in the context of primary acquired melanosis. Approximately 25% of conjunctival melanomas arise from de novo growth.
Histopathology
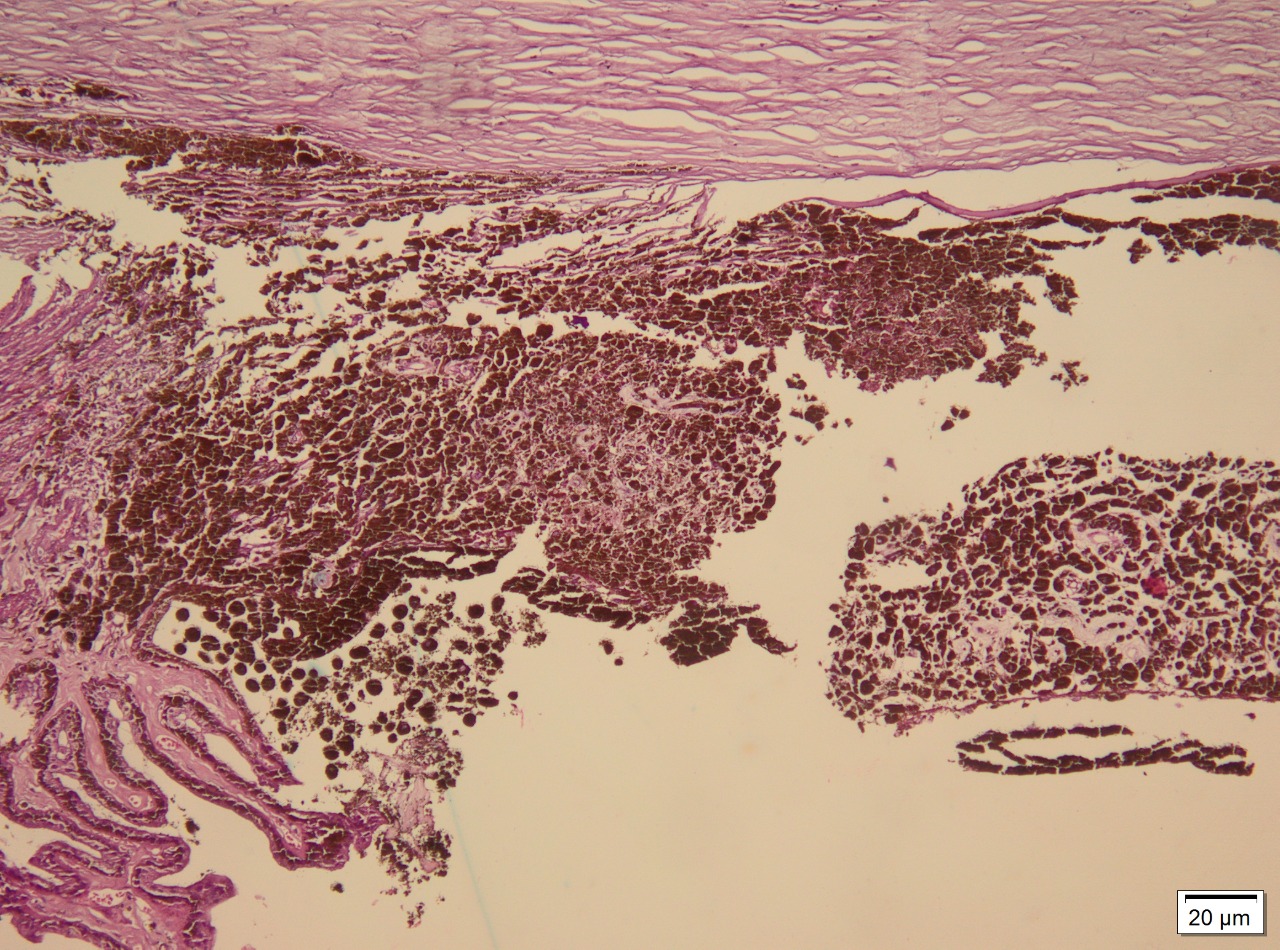
Ocular melanoma exhibits 3 molecular forms: spindle, epithelioid, and mixed. Spindle cells are long and narrow and contain prominent nuclei and nucleoli. Epithelioid cells are larger, resembling epithelial cells, and contain eosinophilic cytoplasm. Occasionally, the tumor may be necrotic and unclassifiable. Spindle cell melanoma accounts for 9%, epithelioid for 5%, and mixed cell for 86%.[9] Staining for S-100, a multigenic family of small acidic EF-hand calcium-binding proteins, is highly sensitive for identifying melanoma. More specific markers for melanoma include immunohistochemical staining for HMB-45, tyrosinase, and melanoma antigen recognized by T cells (MART-1/Melan-A; see Image. Immunohistochemical Staining).[10]
History and Physical
History
Symptoms of ocular melanomas vary based on the tumor's location. Patients often remain asymptomatic, and the malignancy is often detected during routine eye examinations. Choroidal melanomas may stay symptom-free for extended periods, whereas ciliary body tumors remain unnoticed until their growth impacts nearby structures. Iris-affected melanomas might lead to patients noting rapid growth in a nevus present since childhood. Conjunctival lesions can emerge from an unblemished area, a preexisting nevus, or as flat, spreading pigmentation in primary acquired melanosis.
Some patients may experience pain due to increased intraocular pressure or impingement on posterior ciliary nerves. Decreased visual acuity occurs due to cataract formation, tumor growth into the subfoveal retina, cystoid macular edema, retinal detachment, vitreous hemorrhage, and the tumor's direct blockage of the visual axis. Scotoma develops as the perifoveal retina becomes involved. Necrosis within the tumor or adjacent structures produces vitreous hemorrhage or hyphema, leading to floaters.[11]
Physical Examination
Choroidal melanoma: Choroidal melanoma is generally a nodular, dome-shaped, and well-circumscribed mass under the retinal pigment epithelium, adopting the classic mushroom shape with growth. The color varies from amelanotic to darkly pigmented. When the tumor appears lightly colored, the overlying epithelium reveals drusen, atrophy, and orange discoloration. Sentinel vessels may also be visible.
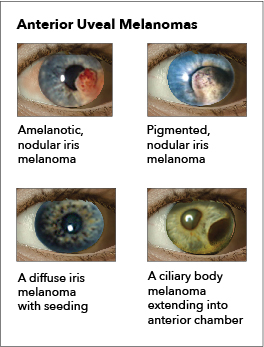
Iris melanoma: Melanomas of the iris are circumscribed or diffuse. Most circumscribed lesions arise in the inferior half of the iris. The color varies between yellow, tan, or brown (see Image. Ocular Melanoma). Patients with a diffuse lesion likely present with a unilateral dark iris.
Ciliary body melanoma: The presence of a sentinel vessel is an early sign of a ciliary body melanoma. This finding should prompt clinicians to dilate a patient's eyes and carefully examine the anterior and posterior segments. In addition, an unexplained, unilateral low intraocular pressure compared to the other healthy eye may also indicate a ciliary body tumor. A difference of 5 mm Hg may be the only indicator of a tumor affecting the ciliary body (see Image. Ocular Melanoma).
Conjunctival melanoma: Conjunctival melanoma tends to become fixed to the underlying tissue and extend onto the peripheral limbus. Some patients may notice pigmentation of the eyelid margins and skin. Lymphadenopathy may also occur in the ipsilateral preauricular, submandibular, and cervical nodes.
Evaluation
The diagnosis of uveal melanoma is typically determined noninvasively through a funduscopic examination and ultrasound. Around 5% of patients require a diagnostic biopsy to confirm the diagnosis. Physical examination and imaging findings that increase the likelihood of malignancy include a tumor thickness greater than 2 mm, subretinal fluid, visual symptoms, acoustic hollowness observed on ultrasound, and a margin within 3 disc diameters of the optic nerve.[12]
Fundus Imaging
The clinician obtains images using an ocular fundus camera and digital photography. Imaging the fundus allows clinicians to determine the size of the tumor, detect retinal detachment, and identify the presence of pigmentation. In addition, clinicians note the tumor location, extrascleral extension anteriorly, and the involvement of the ciliary body. Melanomas appear in dome, mushroom-shaped, or diffuse forms. The color of the tumor typically ranges from gray to greenish-brown.
Ultrasound
Classic ultrasound technology is capable of evaluating tumors of the anterior segment. Clinicians utilize 2 different ultrasound frequencies in the evaluation of ocular melanoma. The A-mode scan is used to obtain measurements or perform a mobility assessment, and a B-mode scan visualizes intraocular structures of different echogenicities. In addition, ultrasound also allows for the assessment of adjacent structures. With its diverse capabilities, ultrasound technology can assess tumors as small as 2 mm and effectively evaluate highly pigmented, nonpigmented, and mean vascular density in tumors.
Ultrasound biomicroscopy uses higher frequencies to evaluate tumors originating from the ciliary body, tumor progression to regions posterior to the iris, and to differentiate between cystic and solid lesions. Ultrasound limitations include potential challenges in detecting lesions <1 mm, the possibility of overestimating tumor size, and the potential for ocular disorders such as choroidal nevi, metastatic neoplasms, and choroidal hemangiomas, which may exhibit similarities to melanoma on ultrasound.
Optical Coherence Tomography
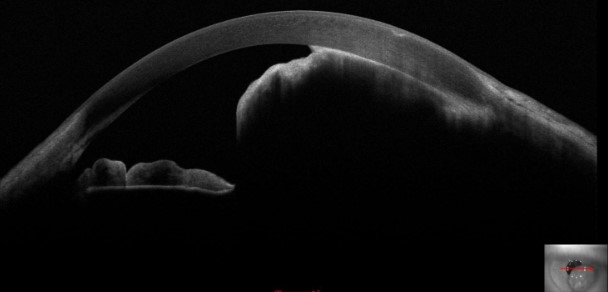
Optical coherence tomography (OCT) involves receiving and processing light waves to build cross-sectional images. Anterior segment OCT allows for real-time imaging of the cornea, anterior chamber angle, sclera, iris, and lens (see Image. Iris Melanoma). Anterior segment OCT detects and differentiates various conditions, including iris melanoma, iris nevi, iris melanocytoma, and ocular surface squamous tumors.[13]
OCT angiography plays a crucial role in the early diagnosis of uveal melanoma and circumscribed choroidal hemangioma. This imaging technique aids in detecting tumor vessels, evaluating their branching patterns, and comparing vessel caliber. The increased ability to identify the tumor's vessels supports early differentiation between malignant and benign tumors. Furthermore, OCT angiography may be a predictive tool for future radiation-induced retinal toxicity and a quantitative endpoint for assessing visual prognosis.
One limitation of OCT is image shadowing, which may occur with pigmented lesions. Because the sclera or iris epithelium limits light penetration, anterior segment OCT is preferred for imaging nonpigmented superficial lesions. OCT provides suboptimal images in tumors with a basal diameter >5 mm, patients older than 60, and extramacular tumors. Experts recommend using OCT for small choroidal tumors and ultrasound for larger tumors.
Fundus Fluorescein Angiography
Fundus fluorescein angiography involves injecting sodium fluorescein dye intravenously to assess circulation in the retina and choroid. This technique captures images by illuminating the retina with blue light, revealing characteristics of the retinal pigment's blood-retina barrier and epithelium. In uveal melanomas, fundus fluorescein angiography detects intrinsic tumor circulation or double circulation, extensive leakage with progressive fluorescence, late staining of the lesion, and multiple pinpoint leaks or hot spots at the level of the retinal pigment epithelium. However, its diagnostic accuracy alone is limited. Fundus fluorescein angiography is advantageous in detecting conditions such as hemangioma, hemorrhage, or choroidal detachment.
Indocyanine green angiography utilizes indocyanine green dye, offering greater visualization of tumor vasculature and distinguishing pigmented and nonpigmented tumors. Fundus autofluorescence involves visualizing stimulated light emission from ocular fluorophores, particularly lipofuscin. Fundus autofluorescence may aid in the early detection of choroidal melanoma and differentiation from benign nevi.
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) allows for tumor localization, assessing tumor dimension and the involvement of adjacent tissues. Limitations include artifacts from blinking and involuntary eye movements, uneven melanin distribution that can contribute to inhomogeneous intensity characteristics, and the inability to distinguish between amelanotic and melanotic melanomas. However, MRI remians useful in detecting metastasis and is an important tool in determining treatment options.
Computed Tomography
Computed tomography (CT) is rarely used to diagnose uveal melanoma. Although CT can assess episcleral and extrascleral growth, it lacks the ability to distinguish uveal melanoma from other conditions and lacks precision in measuring tumor size compared to ultrasound. CT can help confirm the diagnosis, observe bony orbital changes, and aid in metastasis staging, especially in assessing liver and extra-liver metastases.
Positron Emission Tomography and Computed Tomography
Positron emission tomography (PET) combined with CT (PET-CT) detects metabolic abnormalities after the administration of radiotracers such as 18F-fludeoxyglucose (18FDG). The primary role of 18FDG-PET-CT is detecting distant metastases in uveal melanoma, particularly in the liver. PET-CT is valuable for initial staging, confirming hepatic metastases, and assessing early therapy response. Despite its efficacy, PET-CT is an expensive imaging technique associated with a significant radiation dose, necessitating careful consideration of the risk-benefit ratio for individuals diagnosed with uveal melanoma. Uveal melanoma is not consistently 18FDG-avid on PET-CT. Visualizing liver metastases can be challenging due to normal liver uptake of FDG.
Laboratory Evaluation
The most common site for metastasis is the liver. Patients with ocular melanoma should undergo initial laboratory tests, including alkaline phosphatase, glutamic-oxaloacetic transaminase, lactate dehydrogenase, and gamma-glutamyl transpeptidase.
Elevation of liver function tests indicates the need for obtaining an MRI of the liver. Patients with suspected metastatic disease at the time of presentation should also undergo a complete blood count, comprehensive metabolic panel, contrast-enhanced MRI of the liver, and contrast-enhanced CT scan of the chest. Clinicians also offer MRI of the brain to evaluate for metastatic disease of the brain. 18FDG-PET-CT is not initially included in the evaluation. A biopsy is required to confirm distant metastatic lesions.
After tumor removal, genetic testing of the tumor can be helpful for prognosis and post-treatment surveillance of patients with primary uveal melanoma. These chromosomal aberrations can be detected using karyotypes, fluorescent in situ hybridization, and single-nucleotide polymorphism array analysis. Mutations in the BAP1 gene are associated with a high risk of metastatic disease, whereas mutations in SF3B1 and EIF1AX are associated with a more favorable prognosis and late onset of metastatic disease. In addition, patients with primary uveal melanoma should undergo a genotyping assay on whole blood for the presence of human leukocyte antigen (HLA)-A*02:01. Patients with this genotype are eligible for targeted therapy with tebentafusp-tebn.
Treatment / Management
The treatment of ocular melanoma depends on tumor size and location. The treatment options for patients with uveal melanoma include observation, plaque radiation therapy (RT), particle beam radiotherapy, transpupillary thermotherapy, laser photocoagulation, local surgical resection, and enucleation.
Observation is an acceptable option for patients with uveal melanomas who are asymptomatic and have a tumor size <12 mm in diameter and <2 to 3 mm in height. These patients should undergo reevaluation every 2 to 4 months with repeat imaging using fundus photography, ultrasound, OCT, or fundus autofluorescence. Because of the less aggressive nature of melanomas of the iris, these tumors are also often managed with observation. If the tumor is large or rapidly growing, treatment options may include brachytherapy, proton beam RT, local excision, or enucleation, as discussed below.
Radiation Therapy
RT is the most common treatment for primary uveal melanoma and is an eye-salvaging therapy. Uveal melanoma is relatively resistant to radiation, necessitating plaque or charged-particle RT. Plaque brachytherapy utilizes a gold-covered, dish-shaped device with a radioactive source sutured to the eyeball, targeting the tumor. Iodine 125 is the preferred isotope in the United States and emits γ radiation. Following treatment, patients should undergo a regular ophthalmologic examination to monitor for complications.
Charged-particle RT is the second most common form of RT utilized. Plaque brachytherapy and charged-particle RT result in similar local control rates. Charged-particle RT is preferred when the melanoma encircles the optic nerve or if the tumor is too large to accommodate the plaque.
Transpupillary Thermotherapy
Transpupillary thermotherapy, known as diode laser hyperthermia, employs an infrared diode laser and gradually heats the entire tumor to approximately 45 to 60 °C, subsequently inducing deep-penetrating tumor necrosis. With a local recurrence rate of 30% after 3 years, transpupillary thermotherapy is most helpful as an adjunct after RT.
Laser Photocoagulation
Laser photocoagulation utilizes high-temperature thermal energy to damage the tumor. Given the high local recurrence rate and complications, laser photocoagulation is not a frequently used treatment for ocular melanoma.
Local Surgical Resection
Local surgical excision is challenging and associated with severe postoperative complications and high local recurrence rates. Except for conjunctival melanoma, local surgical resection is not a common treatment approach. Clinicians employing this technique typically combine it with adjuvant RT to reduce the risk of local recurrence. The treatment of conjunctival melanoma involves wide local excision followed by double freeze-thaw cryotherapy to the margins, with possible alcohol application.[11] Clinicians may then use adjunctive plaque RT. To prevent tumor cell seeding, incisional biopsy and direct manipulation of the tumor should be avoided.
Enucleation
Enucleation provides no significant survival benefits over plaque brachytherapy in most patients.[14] Surgeons typically reserve enucleation for patients with large tumors, marked extrascleral extension, neovascular glaucoma, extensive retinal detachment, poor visual potential, or for patients who prefer enucleation.
For metastatic uveal melanoma, tebentafusp-tebn, a bispecific glycoprotein 100 peptide-HLA-directed CD3 T-cell engager, increases overall survival. This drug functions as an immune mobilizing monoclonal T-cell receptor against cancer (ImmTAC) class of T-cell–directed therapy and specifically targets HLA-A*02:01-positive metastatic uveal melanoma.[15][16] In addition, darovasertib, an inhibitor of protein kinase C, increases overall survival in patients with metastatic uveal melanoma.[17] Further development of specific immunotherapy for ocular melanoma may lead to an increased frequency of performing biopsies to help direct therapy.
Systemic Surveillance
Molecular prognostic testing stratifies patients based on risk and guides surveillance for metastatic disease. Clinicians categorize patients into low, medium, or high risk.
Low risk: Patients in this category have a lower risk of metastasis. They typically exhibit specific characteristics as follows:
- Being classified as American Joint Committee on Cancer (AJCC) T1 (Refer to the Staging section for more information regarding staging ocular melanoma)
- Disomy 3
- Gain of chromosome 6p
- EIF1AX mutation
Patients with low-risk tumors undergo systemic imaging annually.
Medium risk: Patients categorized as medium risk have an intermediate likelihood of developing metastatic disease. They may have the following characteristics.
- SF3B1 mutation
- AJCC T2 and T3 disease
Patients with medium-risk tumors undergo systemic imaging every 6 to 12 months for 10 years and then as clinically indicated.
High risk: Patients in the high-risk category have the greatest likelihood of metastasis. They may exhibit the following characteristics.
- Monosomy 3
- Gain chromosome 8q
- BAP1 mutation
- PRAME nuclear receptor transcriptional regulator (PRAME) expression
- AJCC T4 disease
- Extraocular extension
- Ciliary body involvement
Patients with high-risk tumors undergo systemic imaging every 3 to 6 months for the first 5 years, followed by every 6 to 12 months for years 6 to 12, and then as clinically indicated. MRI is the preferred diagnostic study for systemic imaging. CT scan and ultrasound are acceptable alternatives for patients with low-risk tumors.
Differential Diagnosis
In a clinical setting, ocular melanoma may exhibit similarities to various diseases. Although distinguishing these growths based on their clinical appearance is possible, a biopsy remains the sole definitive method to confirm a diagnosis. The differential diagnoses for choroidal melanoma and ciliary body melanoma are as follows:
- Choroidal detachment
- Intraocular foreign body
- Chronic angle closure glaucoma
- Hyphema
- Cavernous hemangioma
- Vitreous hemorrhage
- Age-related macular degeneration
- Melanocytoma
- Medulloepithelioma
- Choroidal osteoma
- Adenoma
- Adenocarcinoma
- Combined hamartoma of the retina and pigment epithelium
- Congenital hypertrophy and reactive hyperplasia of the retinal pigment epithelium
- Lymphoid tumor
- Hemangiopericytoma
- Leiomyoma
- Neurofibroma
- Glioneuroma
- Astrocytoma
- Rhabdomyosarcoma
- Posterior uveitis
- Sarcoid nodules
- Tubercular granuloma
- Uveitis
The differential diagnoses for iris melanoma are as follows:
- Iris pigment epithelial cyst
- Iris stromal cyst
- Iris nevi
- Iris metastases
- Iris foreign body
- Iridocorneal endothelial syndrome
- Koeppe or Busacca nodules
- Lisch nodules
- Peripheral anterior synechiae
- Other iris tumors, such as leiomyoma and rhabdomyosarcoma
- Cogan-Reese syndrome
Clinicians must distinguish conjunctival melanoma from other conditions, including conjunctival squamous cell carcinoma; conjunctival melanosis; conjunctival mycosis; conjunctival seborrheic keratosis; acquired melanosis; foreign body like graphite; drug toxicity, such as epinephrine; and pseudomelanoma.
Staging
Clinicians stage ocular melanoma using the Tumor, Node, Metastasis (TNM) system. In this system, T designates the tumor size, ranging from 1 to 4, with 1 indicating a small tumor and 4 indicating a large tumor. N indicates the presence of malignant cells in regional lymph nodes, denoted by either 0 or 1, with 0 representing no spread to the lymph nodes and 1 signifying regional lymph node involvement. M describes the presence of metastatic disease, represented by 0 or 1, with 0 indicating no metastases and 1 indicating the presence of metastases. The eighth edition TNM system appears to be prognostically equivalent to using the largest basal diameter of the tumor for the classification of the ciliary body and choroid tumors.
Prognosis
Local treatment prevents local recurrence in 95% of patients. However, due to micrometastasis, 50% of patients with uveal tumors develop metastatic disease. Metastatic disease is typically fatal, with a median survival of only 10 months after diagnosis.[11][18] Several prognostic indicators leading to poor outcomes include increased patient age, the largest basal diameter of the tumor, ciliary body involvement, extrascleral tumor extension, epithelioid cell type, and vasculogenic mimicry patterns.[19][20]
Chromosomal analysis identifying monosomy 3 and 8q gain can help determine prognosis to some extent, but gene expression profiling proves to have better prognostic abilities. Gene expression profiling assesses the expression of 12 discriminating genes and 3 control genes to determine the metastatic potential, classifying patients into either class 1, indicating low potential, or class 2, indicating high potential. In addition, detecting circulating tumor cells or DNA may be a prognostic indicator for metastatic disease.
The 5-year survival rate for conjunctival melanoma with treatment is 83% to 84%, and the 10-year survival rate is 69% to 80%. The overall mortality for choroidal melanoma and ciliary body melanoma is 30% to 50% within 10 years, primarily due to metastatic disease. The overall mortality rate for iris melanoma is 0% to 11%.
Complications
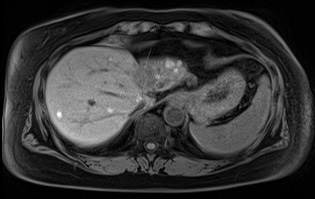
Complications associated with ocular melanoma encompass various issues beyond metastasis and death (see Image. Metastatic Ocular Melanoma). These include vision loss, glaucoma, retinal detachment, cataract formation, astigmatism, and macular edema.[21] Additional complications following RT, potentially emerging up to 5 years after therapy, involve radiation-induced retinopathy and neovascular glaucoma. Intravitreal anti-vascular endothelial growth factor after brachytherapy may mitigate the onset or slow the progression of macular edema and associated vision loss.
Additional post-radiation complications may include dry eye, vitreous hemorrhage, uveitis, scleral necrosis, radiation retinopathy, optic neuropathy, lash loss, and lacrimal gland dysfunction. These adverse effects are more likely to occur with charged-particle RT. In addition to the previously mentioned complications, transpupillary thermotherapy may cause retinal vascular occlusion.
Consultations
Patients suspected or confirmed to have ocular melanoma typically commence their evaluation with an ophthalmologist specializing in ocular oncology. Furthermore, essential consultations include an ocular oncologist, a radiation oncologist, and an occupational therapist. Others may also require psychological support from a psychologist or a psychiatrist.
Deterrence and Patient Education
Despite successful treatment for local disease, ocular melanoma carries a 30% to 50% mortality rate due to metastatic disease. Risk factors for ocular melanoma include fair skin, light-colored eyes, and older age. The American Academy of Ophthalmology recommends a dilated eye examination by age 40 for early detection. Additional risk factors include dysplastic nevus syndrome, a family history of ocular melanoma, eye freckles, nevus of Ota, germline mutation in the BAP1 gene, and occupational exposure to arc welding. Cancer screening is advised for asymptomatic individuals to facilitate early detection and successful treatment. Individuals with predisposing conditions may benefit from annual ophthalmologic examinations. Limiting sunlight exposure could prevent intraocular melanoma in susceptible individuals.
Patients with ocular melanoma should be aware of the risk of metastatic disease years after the initial diagnosis. Monitoring for metastasis should be performed regularly, emphasizing compliance with follow-up visits, regardless of stage and treatment. The frequency of monitoring depends on the tumor stage, and those with a high risk of metastasis should be monitored as frequently as every 3 to 6 months for the first 5 years.[22]
Enhancing Healthcare Team Outcomes
Ocular melanoma is a malignancy characterized by the abnormal growth of melanocytes, representing the second most common type of melanoma and the most common primary intraocular malignancy in adults. Most often originating in the uveal tract, with a minority arising from conjunctival melanocytes, individuals with light-colored irises are at highest risk. Factors such as UV light exposure, genetic predisposition, and certain underlying conditions such as a family history of uveal melanoma, uveal nevus, congenital ocular melanocytosis, dysplastic nevus syndrome, and xeroderma pigmentosum affect the patient's risk.
Detection may occur in an asymptomatic individual incidentally during funduscopic examinations or upon the development of visual symptoms such as decreased acuity, scotoma, or ocular pain. Diagnostic tools include imaging of the fundus, ultrasound, OCT, and MRI, with a biopsy required in a small percentage of cases. Local treatment with RT effectively prevents a recurrence in 95% of patients, but approximately 50% of patients with uveal disease face metastatic risks. Given the disorder's high risk of metastatic disease and associated mortality, understanding the diagnostic process and implementing evidence-based treatment and long-term management strategies are crucial across healthcare specialties.
Genetic testing post-tumor removal aids in prognosis and surveillance, with specific chromosomal aberrations and genetic markers indicating metastatic risk. With the continual development of new specific targeted therapies such as tebentafusp-tebn, indicated for HLA-A*02:01-positive adults, and darovasertib, continued education along with effective interprofessional communication is crucial, allowing seamless information exchange and collaborative decision-making among the team members. By embracing these principles of skill, strategy, interprofessional communication, and care coordination, healthcare professionals can deliver patient-centered care, ultimately improving patient outcomes and enhancing team performance in managing ocular melanoma.