Continuing Education Activity
Ventricular septal defects (VSDs) are the most prevalent congenital cardiac anomaly in children and the second most common in adults, surpassed only by bicuspid aortic valves. The primary mechanism leading to hemodynamic compromise in VSDs is the abnormal communication between the right and left ventricles, resulting in shunt formation.
This activity will focus on understanding the presentation and pathophysiology of VSDs, enabling clinicians to enhance their diagnostic and management skills. Furthermore, the emphasis on interprofessional collaboration underscores the importance of a team-based approach in effectively addressing the complex challenges associated with congenital heart defects. By participating in this activity, clinicians can expect to acquire up-to-date knowledge, practical insights, and strategies for optimizing patient care and outcomes in the context of VSDs.
Objectives:
Differentiate between muscular and non-muscular ventricular septal defects to guide appropriate management strategies and intervention timelines.
Implement systematic screening processes to detect ventricular septal defects early in patients, facilitating timely intervention and improving long-term outcomes.
Select the most appropriate intervention method (surgical or percutaneous closure) based on the type and characteristics of the ventricular septal defects, ensuring personalized and effective patient care.
Collaborate with interprofessional teams, including cardiologists, surgeons, and other specialists, to ensure comprehensive and coordinated care for patients with ventricular septal defects
Introduction
Ventricular septal defects (VSDs) are the most common congenital cardiac anomaly in children and are the second most common congenital abnormality in adults, surpassed only by a bicuspid aortic valve. The primary pathophysiology involves an abnormal communication between the right and left ventricles, leading to shunt formation and subsequent hemodynamic compromise in VSD. While spontaneous closure is observed in many VSD cases, persistent large defects can result in detrimental complications, including pulmonary arterial hypertension (PAH), ventricular dysfunction, and an increased risk of arrhythmias.[1][2][3] VSDs were first identified by Dalrymple in 1847.[4]
Etiology
VSD develops from a developmental abnormality or disruption of the interventricular septum formation during the intricate process of embryologic heart morphogenesis. While VSDs often manifest in isolation, they can also be associated with other congenital heart defects such as atrial septal defects, patent ductus arteriosus, right aortic arch, and pulmonic stenosis. Additionally, VSDs are observed in conditions like aortic coarctation and subaortic stenosis, tetralogy of Fallot, and transposition of the great arteries, contributing to the complexities of congenital heart disease.
Several genetic factors contribute to VSDs, including chromosomal abnormalities, single-gene mutations, and polygenic inheritance. A recently discovered TBX5 mutation has been identified as a cause of septal defects in patients with Holt-Oram syndrome. Noninherited risk factors also play a role in VSD development. Maternal infections (including rubella, influenza, and febrile illness), maternal diabetes mellitus, and phenylketonuria have been implicated. Exposure to toxins like alcohol, marijuana, cocaine, and certain medications such as metronidazole and ibuprofen are also linked to VSDs.[5][6]
Epidemiology
Isolated VSD constitutes 37% of all congenital heart diseases in children, with an incidence of approximately 0.3% among newborns. The occurrence diminishes significantly in adults due to the spontaneous closure observed in up to 90% of cases. VSDs do not exhibit gender predilection.
Pathophysiology
The interventricular septum is an asymmetric curved wall shaped by the pressure disparity in the ventricular chambers. The interventricular septum consists of two distinct parts: a membranous portion and a muscular portion. The membranous part contains the atrioventricular segment, while the muscular portion comprises the trabecular, infundibular, and inlet segments.[7][8]
The failure of development or fusion of any of the above components during morphogenesis of the embryonic heart results in a VSD in the corresponding component. The various anatomic locations and histologic variations of VSDs have given rise to several classifications and nomenclature systems. A new unified classification has been established to streamline the description of VSDs and reduce confusion from multiple synonyms. This classification categorizes VSDs into 4 significant groups:[9]
Infundibular (Outlet) VSD
- Location: Below the semilunar valves (aortic and pulmonary) in the outlet septum of the right ventricle, above the crista supraventricularis.
- Characteristics: Also known as supracristal, it is uncommon, representing only 6% of all VSDs, except in the Asian population, where it can account for approximately 30%. Aortic valve prolapse and regurgitation are common because of loss of support of the right and noncoronary cusps of the aortic valve. It is unusual for these defects to close spontaneously.
Perimembranous VSD
- Location: In the membranous septum, inferior to the crista supraventricularis.
- Characteristics: This is the most common type, accounting for 80% of all VSDs. It may involve the muscular septum and can interchangeably be called membranous VSD. The septal leaflet of the tricuspid valve can form a “pouch,” which can reduce the shunt, resulting in spontaneous closure.
Inlet or Atrioventricular Canal VSD
- Location: Just inferior to the inlet valves (tricuspid and mitral) within the inlet part of the right ventricular septum.
- Characteristics: Represents about 8% of all VSDs and is observed more frequently in patients with Down syndrome.
Muscular (Trabecular) VSD:
- Location: In the muscular septum, usually in the apical, central, and outlet parts of the interventricular septum.
- Characteristics: These VSDs can be multiple, resembling a “Swiss cheese” appearance, representing up to 20% of VSDs in infants. The incidence is lower in adults due to a tendency for spontaneous closure.
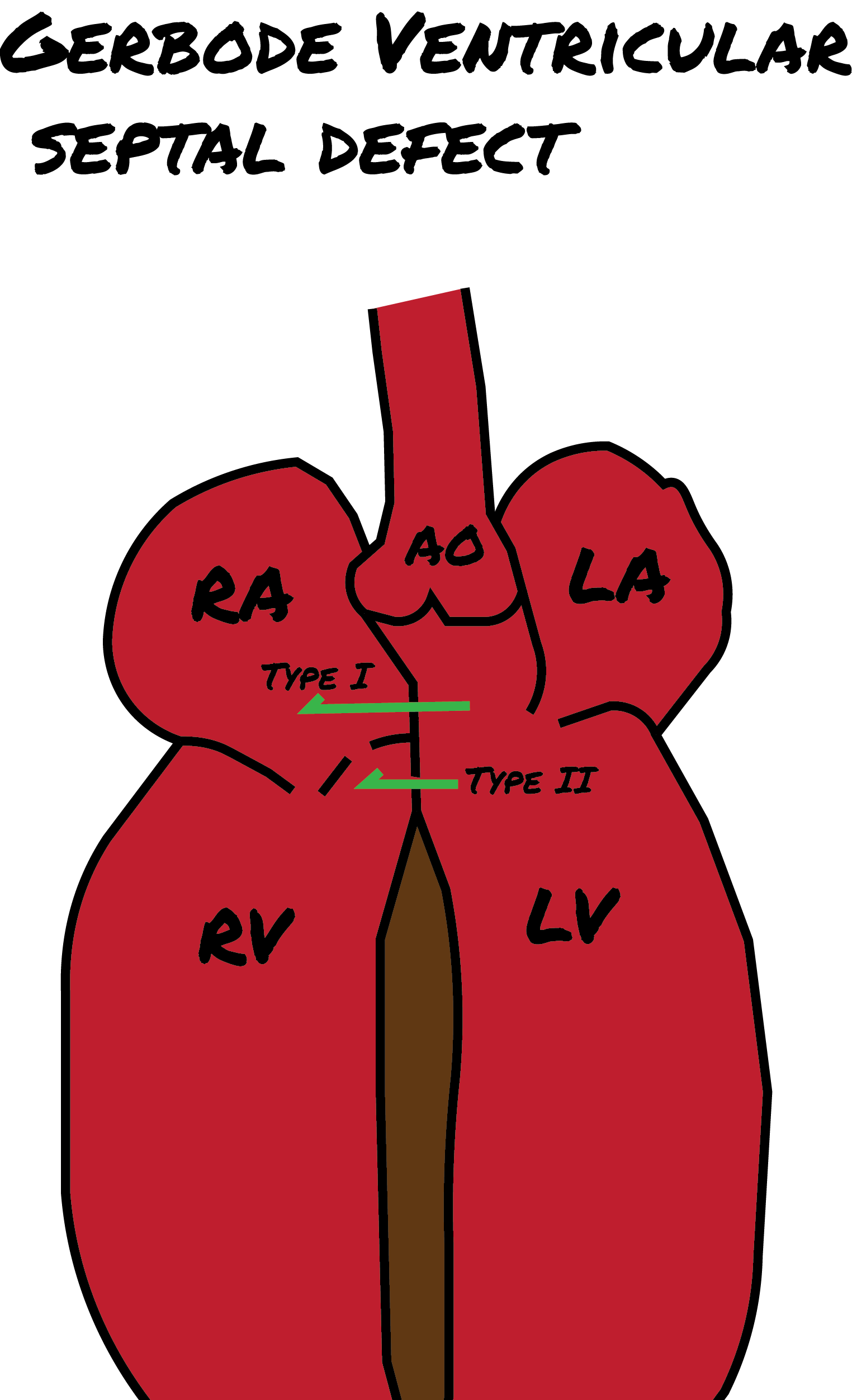
In addition to these 4 types, the Gerbode defect is a distinct type of cardiac anomaly, named after Frank Gerbode et al from Stanford University School of Medicine, who first described it in 1958. This anomaly involves a direct connection between the left ventricle and the right atrium and is classified into 3 forms. (see Image. Gerbode Defect)
- Type I (true gerbode or supravalvular type): This is the rarest type, originating from a congenital deficiency of the atrioventricular component of the membranous septum.
- Type II (subvalvular type): Ventriculo-atrial shunting is caused by a central VSD and a deficiency of the septal leaflet of the tricuspid valve.
- Type III: This type represents a combination of the characteristics observed in Types I and II.[10][11][12]
The primary pathophysiologic mechanism of VSD involves shunt creation between the right and left ventricles. The significance of the VSD in terms of hemodynamics is determined by the amount of blood shunted and the direction of the shunted blood. These factors are influenced by the size and location of the VSD, as well as the pulmonary vascular resistance.
In addition to classifying VSDs based on location, another classification criterion involves categorizing them according to size. The diameter of the aortic annulus characterizes the size. VSDs are considered small if their dimensions are equal to or less than 25% of the aortic annulus diameter, medium if they measure more than 25% but less than 75%, and large if they exceed 75% of the aortic annulus diameter.
In prolonged, large left-to-right shunts, the pulmonary vascular endothelium undergoes irreversible changes, resulting in persistent PAH. When the pressure within the pulmonary circulation surpasses the pressure in the systemic circulation, the shunt direction reverses and becomes a right-to-left shunt. This condition is termed Eisenmenger syndrome and manifests in approximately 10% to 15% of individuals with VSD.
History and Physical
The clinical presentation of unrepaired VSDs primarily depends on the presence of a hemodynamically significant shunt, directly correlating with the size of the defect. VSDs can be described as follows:
- Small VSDs: These defects result in minimal left-to-right shunt without causing left ventricular fluid overload or PAH. As a result, individuals with small VSDs are often asymptomatic or the condition is incidentally discovered during physical examaminations.
- Medium-size VSDs: Individuals with medium-sized VSDs experience moderate left ventricular volume overload and no or mild PAH. Presentation typically occurs late in childhood, accompanied by mild congestive heart failure (CHF).
- Large VSD: Those with significant defects develop CHF early in childhood due to severe left ventricular overload and severe PAH.
The murmur associated with VSD is typically pansystolic and is best heard in the left lower sternal border. The murmur is harsh and loud in small defects, while in large defects, it tends to be softer and less intense. Handgrips increase afterload, intensifying the murmur. Murmurs associated with infundibular defects are best heard in the pulmonic area. A diastolic decrescendo murmur and wide pulse pressure can be detected in aortic regurgitation. Increased left ventricular flow can result in the middiastolic rumble in the lower left sternal border. Sometimes, a septal aneurysm can cause a systolic click, particularly in membranous defects.
In cases of Eisenmenger syndrome, manifestations include cyanosis, desaturation, dyspnea, syncope, secondary erythrocytosis, and clubbing. In such cases, the typical murmur of VSD may be absent, and an accentuated pulmonic component of the second heart sound may be audible.
Evaluation
Color Doppler transthoracic echocardiography (TTE) is the most valuable diagnostic tool for VSDs due to its high sensitivity. Color Doppler TTE can detect up to 95% of VSDs, especially nonapical lesions larger than 5 mm. This imaging technique offers essential morphologic information, including size, location, and the number of defects, along with hemodynamic information, such as jet size, severity, and estimation of pulmonary artery pressure. Additionally, TTE is useful in detecting associated conditions such as aortic insufficiency and other congenital heart defects. TTE also proves useful for assessing right and left ventricular chamber size and function. However, limitations include operator dependence and challenges posed by poor acoustic windows. In cases where conventional TTE results are inconclusive, a transesophageal echo (TEE) is recommended for further evaluation.[13][14]
Electrocardiography (ECG) results appear entirely normal in half of the patients with VSD. When the ECG is abnormal, it may detect left ventricular hypertrophy in those with large shunts. In patients with PAH, the ECG may show right bundle branch block, right axis deviation, and right ventricular hypertrophy and strain.
Chest radiography typically appears normal in individuals with small defects. However, an enlarged cardiac silhouette may be evident in larger defects, accompanied by increased left ventricular size. Those with PAH may exhibit signs of right ventricular enlargement, and an increased pulmonary diameter can be observed in those with PAH.
Cardiac magnetic resonance imaging and computed tomography are helpful in cases where anatomy is complex, such as VSD, accompanied by other congenital heart anomalies and defects in unusual locations that are hard to visualize by conventional TTE.
Cardiac catheterization is a crucial diagnostic tool that offers precise hemodynamic information, especially regarding pulmonary vascular resistance and response to vasodilators. This information is valuable for individuals undergoing evaluation for surgical closure of VSDs. Additionally, cardiac catheterization provides in-depth details about coexisting conditions, such as aortic regurgitation, and proves beneficial in cases involving multiple VSDs or when coronary artery disease is suspected.
Treatment / Management
Approximately 85% to 90% of small isolated VSDs undergo spontaneous closure within the first year of life. Patients with small, asymptomatic VSDs, in the absence of PAH, exhibit an excellent prognosis without intervention. However, if intervention is warranted, the management approach involves VSD closure.
In the case of Eisenmenger syndrome, management is usually conducted in advanced centers due to the inherent complexity of these cases. Historically, surgical repair was the only option; however, recent advances in interventional techniques enable percutaneous VSD closure.
It is no longer recommended to routinely prescribe antibiotic prophylaxis for infective endocarditis in patients with unrepaired VSDs.[15] Endocarditis prophylaxis is mainly indicated in cyanotic congenital heart disease, prior endocarditis episodes, and in individuals with prosthetic heart valves or those who have undergone repair with prosthetic material.
Generally, VSD closure is indicated in medium to large defects causing significant hemodynamic compromise, especially in symptomatic cases with left ventricular dysfunction. Intervention may also be considered in cases of progressive aortic insufficiency or following an episode of endocarditis. Indications for surgical closure of VSDs, according to the American College of Cardiology and American Heart Association 2008 guidelines, include:
- Pulmonary to systemic blood flow (Qp/Qs) ratio of ≥2 with clinical evidence of left ventricular fluid overload
- Intervention may be considered for milder shunts (Qp/Qs >1.5) if there is evidence of left ventricular systolic or diastolic dysfunction. Additionally, surgical closure is reasonable when the pulmonary artery pressure and pulmonary vascular resistance are less than two-thirds of systemic pressure and systemic vascular resistance, respectively.
Surgical repair of VSDs offers several benefits, including a reduced risk of endocarditis, potential improvement in PAH, and overall survival. In the absence of PAH, the operative mortality rate is approximately 1%. However, surgical intervention is not without potential complications.
Complications of VSD surgical repair include:
- Residual or recurrent VSD
- Valvular incompetence: Tricuspid regurgitation and aortic insufficiency
- Arrhythmias: common postoperative arrhythmias include atrial fibrillation, complete heart block, and ventricular tachycardia
- Left ventricular dysfunction
The main contraindication for surgical VSD closure is the presence of irreversible PAH. This is due to the high perioperative mortality and the potential for significant pulmonary complications in this subset of patients.
Percutaneous device VSD closure is reserved for individuals for whom surgery poses a high risk due to factors such as severe PAH, multiple comorbidities, or a history of prior cardiothoracic surgery, including cases with residual or recurrent VSD. This procedure is particularly suitable for muscular VSDs, but its applicability to cases involving defects near the inlet valves can be challenging.
While percutaneous VSD closure remains relatively unpopular in the US, current data indicates excellent outcomes with high rates of complete closure and low mortality. The most frequent complication associated with this procedure is a complete atrioventricular block, primarily observed in perimembranous defects.
Closure of Gerbode defects is generally recommended to prevent the progression of tricuspid regurgitation and the potential occurrence of infective endocarditis. While it is uncommon for Gerbode defects to close spontaneously, effective treatment options exist.
- Surgical closure is the most common and widely employed treatment for Gerbode defects. It is a well-established procedure with favorable outcomes.
- Transcatheter device closure is an effective alternative treatment. Although it is less common, there have been reports of successful transcatheter closure for both congenital and acquired Gerbode defects. However, there are considerations during this procedure, including ensuring proper alignment of the device, potential conduction disturbances, and the possibility of exacerbating tricuspid regurgitation.
In summary, VSD is the most common congenital anomaly observed at birth. While small defects are often anticipated to close spontaneously within the first year of life, larger defects can result in severe complications. Surgical VSD closure and device closure are the primary interventions recommended for addressing large defects.[16][17][18]
Differential Diagnosis
While VSD is a specific condition, several other cardiac and noncardiac conditions may present with similar symptoms. The differential diagnosis for VSD includes:
- Atrioventricular septal defect
- Atrial septal defect
- Patent ductus arteriosus
- Pulmonary stenosis
- Tetralogy of Fallot
- Mitral valve prolapse
- Tricuspid regurgitation
- Eisenmenger syndrome
- Infective endocarditisis
- Myocarditis
Prognosis
Patients who have undergone VSD repair generally have a favorable prognosis. Nonetheless, in the long run, they carry an elevated risk of arrhythmia, endocarditis, and CHF compared to the general population.[19]
Complications
Complications of VSD may include:
- Eisenmenger syndrome
- Aortic insufficiency due to prolapse of the aortic valve leaflet
- Endocarditis
- Embolization
- CHF
- Pulmonary hypertension
- Arrythmias
Consultations
For the evaluation and management of VSD, consultations with the following specialists may be necessary:
- Cardiologist (pediatric and adult)
- Cardiothoracic surgeon
- Cardiac electrophysiologist
Deterrence and Patient Education
Parents of patients with small Ventricular Septal Defects (VSDs) should be informed that, often, neither medical nor surgical intervention is necessary as these defects commonly close over time. Prophylactic antibiotics to prevent endocarditis are typically no longer mandated. However, promoting good oral hygiene remains essential to minimize the risk of endocarditis.[15] Adherence to medications and close follow-up with specialists is essential for all patients with VSDs.
Pearls and Other Issues
Key facts to keep in mind about VSD are as follows:
- Many small VSDs, especially muscular ones, may close spontaneously within the first year of life.
- The pansystolic murmur associated with VSD may be mistaken for the pansystolic murmur of mitral regurgitation. The VSD murmur intensifies toward the sternum, whereas the murmur of mitral regurgitation increases in intensity away from the sternum.
- Prophylactic antibiotics to prevent endocarditis are generally no longer recommended in most cases.
- Larger or unrepaired VSDs can lead to complications such as arrhythmias, congestive heart failure, and endocarditis.
- While antibiotics may not be required, maintaining good oral hygiene is crucial to reduce the risk of endocarditis.
Enhancing Healthcare Team Outcomes
The optimal management of VSD involves a collaborative interprofessional team, including a pediatrician, cardiologist, cardiac surgeon, nurses (intensive care and cardiac), physical therapist, and social worker. Parent and patient education is crucial for promoting follow-up care.
Some individuals with perimembranous VSD may develop aortic valve prolapse, necessitating surgical intervention. Unrepaired VSDs carry the risk of increased pulmonary vascular resistance, leading to Eisenmenger syndrome. Unfortunately, aside from heart and lung transplantation, viable therapies are limited. Due to organ shortages, many patients with Eisenenger syndrome face progressive right heart failure and cyanosis, often resulting in unfavorable outcomes.[20][21]
Young, asymptomatic children with small VSDs typically experience favorable outcomes. However, symptoms may manifest if complications like anemia, infection, or endocarditis arise. In contrast, individuals with large, unrepaired VSDs face poor outcomes due to contained left-to-right shunting, culminating in pulmonary hypertension and Eisenmenger syndrome. Elective VSD repair in infants within the first 2 years of life is standard in North America, yielding a mortality of less than 1% and allowing most patients to enjoy an average lifespan.[22]