[1]
STEELE JC, RICHARDSON JC, OLSZEWSKI J. PROGRESSIVE SUPRANUCLEAR PALSY. A HETEROGENEOUS DEGENERATION INVOLVING THE BRAIN STEM, BASAL GANGLIA AND CEREBELLUM WITH VERTICAL GAZE AND PSEUDOBULBAR PALSY, NUCHAL DYSTONIA AND DEMENTIA. Archives of neurology. 1964 Apr:10():333-59
[PubMed PMID: 14107684]
[3]
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Current opinion in neurology. 2010 Aug:23(4):394-400. doi: 10.1097/WCO.0b013e32833be924. Epub
[PubMed PMID: 20610990]
Level 3 (low-level) evidence
[4]
Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, McKee A, Dickson D, Bancher C, Tabaton M, Jellinger K, Anderson DW. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. Journal of neuropathology and experimental neurology. 1996 Jan:55(1):97-105
[PubMed PMID: 8558176]
[5]
Golbe LI, Davis PH, Schoenberg BS, Duvoisin RC. Prevalence and natural history of progressive supranuclear palsy. Neurology. 1988 Jul:38(7):1031-4
[PubMed PMID: 3386818]
[6]
Golbe LI, Rubin RS, Cody RP, Belsh JM, Duvoisin RC, Grosmann C, Lepore FE, Mark MH, Sachdeo RC, Sage JI, Zimmerman TR Jr. Follow-up study of risk factors in progressive supranuclear palsy. Neurology. 1996 Jul:47(1):148-54
[PubMed PMID: 8710069]
[7]
Litvan I, Lees PS, Cunningham CR, Rai SN, Cambon AC, Standaert DG, Marras C, Juncos J, Riley D, Reich S, Hall D, Kluger B, Bordelon Y, Shprecher DR, for ENGENE-PSP. Environmental and occupational risk factors for progressive supranuclear palsy: Case-control study. Movement disorders : official journal of the Movement Disorder Society. 2016 May:31(5):644-52. doi: 10.1002/mds.26512. Epub 2016 Feb 8
[PubMed PMID: 26854325]
Level 2 (mid-level) evidence
[8]
Kawashima M, Miyake M, Kusumi M, Adachi Y, Nakashima K. Prevalence of progressive supranuclear palsy in Yonago, Japan. Movement disorders : official journal of the Movement Disorder Society. 2004 Oct:19(10):1239-40
[PubMed PMID: 15390010]
[9]
Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet (London, England). 1999 Nov 20:354(9192):1771-5
[PubMed PMID: 10577638]
Level 2 (mid-level) evidence
[10]
Rajput AH, Offord KP, Beard CM, Kurland LT. Epidemiology of parkinsonism: incidence, classification, and mortality. Annals of neurology. 1984 Sep:16(3):278-82
[PubMed PMID: 6333204]
[11]
Radhakrishnan K, Thacker AK, Maloo JC, Gerryo SE, Mousa ME. Descriptive epidemiology of some rare neurological diseases in Benghazi, Libya. Neuroepidemiology. 1988:7(3):159-64
[PubMed PMID: 3405368]
[12]
Mastaglia FL, Grainger K, Kee F, Sadka M, Lefroy R. Progressive supranuclear palsy (the Steele-Richardson-Olszewski syndrome) clinical and electrophysiological observations in eleven cases. Proceedings of the Australian Association of Neurologists. 1973:10(0):35-44
[PubMed PMID: 4792160]
Level 3 (low-level) evidence
[13]
Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology. 1997 Nov:49(5):1284-8
[PubMed PMID: 9371909]
[14]
Wenning GK, Litvan I, Tolosa E. Milestones in atypical and secondary Parkinsonisms. Movement disorders : official journal of the Movement Disorder Society. 2011 May:26(6):1083-95. doi: 10.1002/mds.23713. Epub
[PubMed PMID: 21626553]
[15]
Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ, van Swieten JC, Troakes C, Al Sarraj S, Gelpi E, Gaig C, Tolosa E, Oertel WH, Giese A, Roeber S, Arzberger T, Wagenpfeil S, Höglinger GU, Movement Disorder Society-endorsed PSP Study Group. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Movement disorders : official journal of the Movement Disorder Society. 2014 Dec:29(14):1758-66. doi: 10.1002/mds.26054. Epub 2014 Nov 5
[PubMed PMID: 25370486]
Level 2 (mid-level) evidence
[16]
Coyle-Gilchrist IT, Dick KM, Patterson K, Vázquez Rodríquez P, Wehmann E, Wilcox A, Lansdall CJ, Dawson KE, Wiggins J, Mead S, Brayne C, Rowe JB. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016 May 3:86(18):1736-43. doi: 10.1212/WNL.0000000000002638. Epub 2016 Apr 1
[PubMed PMID: 27037234]
[17]
Respondek G, Kurz C, Arzberger T, Compta Y, Englund E, Ferguson LW, Gelpi E, Giese A, Irwin DJ, Meissner WG, Nilsson C, Pantelyat A, Rajput A, van Swieten JC, Troakes C, Josephs KA, Lang AE, Mollenhauer B, Müller U, Whitwell JL, Antonini A, Bhatia KP, Bordelon Y, Corvol JC, Colosimo C, Dodel R, Grossman M, Kassubek J, Krismer F, Levin J, Lorenzl S, Morris H, Nestor P, Oertel WH, Rabinovici GD, Rowe JB, van Eimeren T, Wenning GK, Boxer A, Golbe LI, Litvan I, Stamelou M, Höglinger GU, Movement Disorder Society-Endorsed PSP Study Group. Which ante mortem clinical features predict progressive supranuclear palsy pathology? Movement disorders : official journal of the Movement Disorder Society. 2017 Jul:32(7):995-1005. doi: 10.1002/mds.27034. Epub 2017 May 13
[PubMed PMID: 28500752]
[18]
Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, Mollenhauer B, Müller U, Nilsson C, Whitwell JL, Arzberger T, Englund E, Gelpi E, Giese A, Irwin DJ, Meissner WG, Pantelyat A, Rajput A, van Swieten JC, Troakes C, Antonini A, Bhatia KP, Bordelon Y, Compta Y, Corvol JC, Colosimo C, Dickson DW, Dodel R, Ferguson L, Grossman M, Kassubek J, Krismer F, Levin J, Lorenzl S, Morris HR, Nestor P, Oertel WH, Poewe W, Rabinovici G, Rowe JB, Schellenberg GD, Seppi K, van Eimeren T, Wenning GK, Boxer AL, Golbe LI, Litvan I, Movement Disorder Society-endorsed PSP Study Group. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Movement disorders : official journal of the Movement Disorder Society. 2017 Jun:32(6):853-864. doi: 10.1002/mds.26987. Epub 2017 May 3
[PubMed PMID: 28467028]
[19]
Santacruz P, Uttl B, Litvan I, Grafman J. Progressive supranuclear palsy: a survey of the disease course. Neurology. 1998 Jun:50(6):1637-47
[PubMed PMID: 9633705]
Level 3 (low-level) evidence
[20]
De Bruin VM, Lees AJ. Subcortical neurofibrillary degeneration presenting as Steele-Richardson-Olszewski and other related syndromes: a review of 90 pathologically verified cases. Movement disorders : official journal of the Movement Disorder Society. 1994 Jul:9(4):381-9
[PubMed PMID: 7969203]
Level 3 (low-level) evidence
[21]
Kristensen MO. Progressive supranuclear palsy--20 years later. Acta neurologica Scandinavica. 1985 Mar:71(3):177-89
[PubMed PMID: 3993325]
[22]
Kovacs GG. Invited review: Neuropathology of tauopathies: principles and practice. Neuropathology and applied neurobiology. 2015 Feb:41(1):3-23. doi: 10.1111/nan.12208. Epub
[PubMed PMID: 25495175]
[23]
Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. Journal of neurology. 1999 Sep:246 Suppl 2():II6-15
[PubMed PMID: 10525997]
[24]
Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994 Nov:44(11):2015-9
[PubMed PMID: 7969952]
[25]
Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, Goetz CG, Golbe LI, Grafman J, Growdon JH, Hallett M, Jankovic J, Quinn NP, Tolosa E, Zee DS. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul:47(1):1-9
[PubMed PMID: 8710059]
[26]
Birdi S, Rajput AH, Fenton M, Donat JR, Rozdilsky B, Robinson C, Macaulay R, George D. Progressive supranuclear palsy diagnosis and confounding features: report on 16 autopsied cases. Movement disorders : official journal of the Movement Disorder Society. 2002 Nov:17(6):1255-64
[PubMed PMID: 12465065]
Level 3 (low-level) evidence
[27]
Chu FC, Reingold DB, Cogan DG, Williams AC. The eye movement disorders of progressive supranuclear palsy. Ophthalmology. 1979 Mar:86(3):422-8
[PubMed PMID: 530592]
[28]
Chen AL, Riley DE, King SA, Joshi AC, Serra A, Liao K, Cohen ML, Otero-Millan J, Martinez-Conde S, Strupp M, Leigh RJ. The disturbance of gaze in progressive supranuclear palsy: implications for pathogenesis. Frontiers in neurology. 2010:1():147. doi: 10.3389/fneur.2010.00147. Epub 2010 Dec 3
[PubMed PMID: 21188269]
[29]
Pierrot-Deseilligny C, Rivaud S, Pillon B, Fournier E, Agid Y. Lateral visually-guided saccades in progressive supranuclear palsy. Brain : a journal of neurology. 1989 Apr:112 ( Pt 2)():471-87
[PubMed PMID: 2706440]
[30]
Litvan I. Update on progressive supranuclear palsy. Current neurology and neuroscience reports. 2004 Jul:4(4):296-302
[PubMed PMID: 15217544]
[31]
Boeve BF. Progressive supranuclear palsy. Parkinsonism & related disorders. 2012 Jan:18 Suppl 1():S192-4. doi: 10.1016/S1353-8020(11)70060-8. Epub
[PubMed PMID: 22166432]
[32]
Robbins TW, James M, Owen AM, Lange KW, Lees AJ, Leigh PN, Marsden CD, Quinn NP, Summers BA. Cognitive deficits in progressive supranuclear palsy, Parkinson's disease, and multiple system atrophy in tests sensitive to frontal lobe dysfunction. Journal of neurology, neurosurgery, and psychiatry. 1994 Jan:57(1):79-88
[PubMed PMID: 8301310]
[33]
Golbe LI, Boeve BF, Keegan BM, Parisi JE. An 81-year-old man with imbalance and memory impairment. Neurology. 2007 Apr 3:68(14):1147-52
[PubMed PMID: 17404198]
[34]
Pillon B, Dubois B, Lhermitte F, Agid Y. Heterogeneity of cognitive impairment in progressive supranuclear palsy, Parkinson's disease, and Alzheimer's disease. Neurology. 1986 Sep:36(9):1179-85
[PubMed PMID: 3748383]
[35]
Dubois B, Pillon B, Legault F, Agid Y, Lhermitte F. Slowing of cognitive processing in progressive supranuclear palsy. A comparison with Parkinson's disease. Archives of neurology. 1988 Nov:45(11):1194-9
[PubMed PMID: 3190499]
[36]
Pharr V, Uttl B, Stark M, Litvan I, Fantie B, Grafman J. Comparison of apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology. 2001 Apr 10:56(7):957-63
[PubMed PMID: 11294936]
[37]
Litvan I, Mega MS, Cummings JL, Fairbanks L. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996 Nov:47(5):1184-9
[PubMed PMID: 8909427]
[38]
Schrag A, Sheikh S, Quinn NP, Lees AJ, Selai C, Mathias C, Litvan I, Lang AE, Bower JH, Burn DJ, Low P, Jahanshahi M. A comparison of depression, anxiety, and health status in patients with progressive supranuclear palsy and multiple system atrophy. Movement disorders : official journal of the Movement Disorder Society. 2010 Jun 15:25(8):1077-81. doi: 10.1002/mds.22794. Epub
[PubMed PMID: 20535826]
[39]
Fukui T, Lee E, Hosoda H, Okita K. Obsessive-compulsive behavior as a symptom of dementia in progressive supranuclear palsy. Dementia and geriatric cognitive disorders. 2010:30(2):179-88. doi: 10.1159/000310351. Epub 2010 Aug 26
[PubMed PMID: 20798538]
[40]
Boeve BF, Silber MH, Parisi JE, Dickson DW, Ferman TJ, Benarroch EE, Schmeichel AM, Smith GE, Petersen RC, Ahlskog JE, Matsumoto JY, Knopman DS, Schenck CH, Mahowald MW. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003 Jul 8:61(1):40-5
[PubMed PMID: 12847154]
[41]
Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, Holton JL, Revesz T, Lees AJ. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson's syndrome and PSP-parkinsonism. Brain : a journal of neurology. 2005 Jun:128(Pt 6):1247-58
[PubMed PMID: 15788542]
[42]
Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ, Revesz T. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson's syndrome. Brain : a journal of neurology. 2007 Jun:130(Pt 6):1566-76
[PubMed PMID: 17525140]
[43]
Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Movement disorders : official journal of the Movement Disorder Society. 2007 Nov 15:22(15):2235-41
[PubMed PMID: 17712855]
[44]
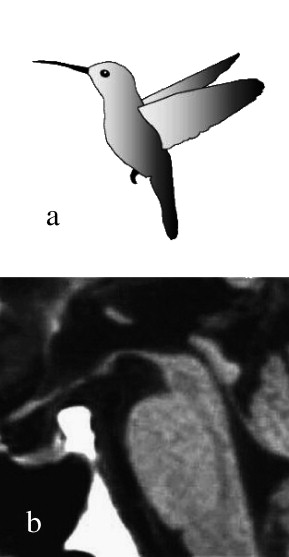
Kato N, Arai K, Hattori T. Study of the rostral midbrain atrophy in progressive supranuclear palsy. Journal of the neurological sciences. 2003 Jun 15:210(1-2):57-60
[PubMed PMID: 12736089]
[45]
Massey LA, Jäger HR, Paviour DC, O'Sullivan SS, Ling H, Williams DR, Kallis C, Holton J, Revesz T, Burn DJ, Yousry T, Lees AJ, Fox NC, Micallef C. The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology. 2013 May 14:80(20):1856-61. doi: 10.1212/WNL.0b013e318292a2d2. Epub 2013 Apr 24
[PubMed PMID: 23616165]
[46]
Kägi G, Bhatia KP, Tolosa E. The role of DAT-SPECT in movement disorders. Journal of neurology, neurosurgery, and psychiatry. 2010 Jan:81(1):5-12. doi: 10.1136/jnnp.2008.157370. Epub
[PubMed PMID: 20019219]
[47]
Kepe V, Bordelon Y, Boxer A, Huang SC, Liu J, Thiede FC, Mazziotta JC, Mendez MF, Donoghue N, Small GW, Barrio JR. PET imaging of neuropathology in tauopathies: progressive supranuclear palsy. Journal of Alzheimer's disease : JAD. 2013:36(1):145-53. doi: 10.3233/JAD-130032. Epub
[PubMed PMID: 23579330]
[48]
Stamelou M, Höglinger G. A Review of Treatment Options for Progressive Supranuclear Palsy. CNS drugs. 2016 Jul:30(7):629-36. doi: 10.1007/s40263-016-0347-2. Epub
[PubMed PMID: 27222018]
[49]
Golbe LI, Ohman-Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain : a journal of neurology. 2007 Jun:130(Pt 6):1552-65
[PubMed PMID: 17405767]
[50]
Sale P, Castiglioni D, De Pandis MF, Torti M, Dall'armi V, Radicati FG, Stocchi F. The Lee Silverman Voice Treatment (LSVT®) speech therapy in progressive supranuclear palsy. European journal of physical and rehabilitation medicine. 2015 Oct:51(5):569-74
[PubMed PMID: 26138088]
[51]
Scelzo E, Lozano AM, Hamani C, Poon YY, Aldakheel A, Zadikoff C, Lang AE, Moro E. Peduncolopontine nucleus stimulation in progressive supranuclear palsy: a randomised trial. Journal of neurology, neurosurgery, and psychiatry. 2017 Jul:88(7):613-616. doi: 10.1136/jnnp-2016-315192. Epub 2017 Feb 18
[PubMed PMID: 28214797]
Level 1 (high-level) evidence
[52]
Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN, NNIPPS Study Group. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain : a journal of neurology. 2009 Jan:132(Pt 1):156-71. doi: 10.1093/brain/awn291. Epub 2008 Nov 23
[PubMed PMID: 19029129]
[53]
Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, Doody RS, Lees A, Golbe LI, Williams DR, Corvol JC, Ludolph A, Burn D, Lorenzl S, Litvan I, Roberson ED, Höglinger GU, Koestler M, Jack CR Jr, Van Deerlin V, Randolph C, Lobach IV, Heuer HW, Gozes I, Parker L, Whitaker S, Hirman J, Stewart AJ, Gold M, Morimoto BH, AL-108-231 Investigators. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. The Lancet. Neurology. 2014 Jul:13(7):676-85. doi: 10.1016/S1474-4422(14)70088-2. Epub 2014 May 27
[PubMed PMID: 24873720]
Level 1 (high-level) evidence
[54]
Tolosa E, Litvan I, Höglinger GU, Burn D, Lees A, Andrés MV, Gómez-Carrillo B, León T, Del Ser T, TAUROS Investigators. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Movement disorders : official journal of the Movement Disorder Society. 2014 Apr:29(4):470-8. doi: 10.1002/mds.25824. Epub 2014 Feb 14
[PubMed PMID: 24532007]
[55]
Leclair-Visonneau L, Rouaud T, Debilly B, Durif F, Houeto JL, Kreisler A, Defebvre L, Lamy E, Volteau C, Nguyen JM, Dily SL, Damier P, Boutoleau-Bretonnière C, Lejeune P, Derkinderen P. Randomized placebo-controlled trial of sodium valproate in progressive supranuclear palsy. Clinical neurology and neurosurgery. 2016 Jul:146():35-9. doi: 10.1016/j.clineuro.2016.04.021. Epub 2016 Apr 27
[PubMed PMID: 27136096]
Level 1 (high-level) evidence
[56]
Nuebling G, Hensler M, Paul S, Zwergal A, Crispin A, Lorenzl S. PROSPERA: a randomized, controlled trial evaluating rasagiline in progressive supranuclear palsy. Journal of neurology. 2016 Aug:263(8):1565-74. doi: 10.1007/s00415-016-8169-1. Epub 2016 May 26
[PubMed PMID: 27230855]
Level 1 (high-level) evidence
[57]
Apetauerova D, Scala SA, Hamill RW, Simon DK, Pathak S, Ruthazer R, Standaert DG, Yacoubian TA. CoQ10 in progressive supranuclear palsy: A randomized, placebo-controlled, double-blind trial. Neurology(R) neuroimmunology & neuroinflammation. 2016 Oct:3(5):e266. doi: 10.1212/NXI.0000000000000266. Epub 2016 Aug 2
[PubMed PMID: 27583276]
Level 1 (high-level) evidence
[58]
Stamelou M, Reuss A, Pilatus U, Magerkurth J, Niklowitz P, Eggert KM, Krisp A, Menke T, Schade-Brittinger C, Oertel WH, Höglinger GU. Short-term effects of coenzyme Q10 in progressive supranuclear palsy: a randomized, placebo-controlled trial. Movement disorders : official journal of the Movement Disorder Society. 2008 May 15:23(7):942-949. doi: 10.1002/mds.22023. Epub
[PubMed PMID: 18464278]
Level 1 (high-level) evidence
[59]
O'Sullivan SS, Massey LA, Williams DR, Silveira-Moriyama L, Kempster PA, Holton JL, Revesz T, Lees AJ. Clinical outcomes of progressive supranuclear palsy and multiple system atrophy. Brain : a journal of neurology. 2008 May:131(Pt 5):1362-72. doi: 10.1093/brain/awn065. Epub 2008 Apr 2
[PubMed PMID: 18385183]
Level 2 (mid-level) evidence
[60]
Testa D, Monza D, Ferrarini M, Soliveri P, Girotti F, Filippini G. Comparison of natural histories of progressive supranuclear palsy and multiple system atrophy. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2001 Jun:22(3):247-51
[PubMed PMID: 11731878]
[61]
dell'Aquila C, Zoccolella S, Cardinali V, de Mari M, Iliceto G, Tartaglione B, Lamberti P, Logroscino G. Predictors of survival in a series of clinically diagnosed progressive supranuclear palsy patients. Parkinsonism & related disorders. 2013 Nov:19(11):980-5. doi: 10.1016/j.parkreldis.2013.06.014. Epub 2013 Aug 19
[PubMed PMID: 23968651]
[62]
Pekmezović T, Ječmenica-Lukić M, Petrović I, Špica V, Tomić A, Kostić VS. Quality of life in patients with progressive supranuclear palsy: one-year follow-up. Journal of neurology. 2015 Sep:262(9):2042-8. doi: 10.1007/s00415-015-7815-3. Epub 2015 Jun 13
[PubMed PMID: 26070289]
Level 2 (mid-level) evidence
[63]
Glasmacher SA, Leigh PN, Saha RA. Predictors of survival in progressive supranuclear palsy and multiple system atrophy: a systematic review and meta-analysis. Journal of neurology, neurosurgery, and psychiatry. 2017 May:88(5):402-411. doi: 10.1136/jnnp-2016-314956. Epub 2017 Mar 1
[PubMed PMID: 28250027]
Level 1 (high-level) evidence
[64]
Clerici I, Ferrazzoli D, Maestri R, Bossio F, Zivi I, Canesi M, Pezzoli G, Frazzitta G. Rehabilitation in progressive supranuclear palsy: Effectiveness of two multidisciplinary treatments. PloS one. 2017:12(2):e0170927. doi: 10.1371/journal.pone.0170927. Epub 2017 Feb 3
[PubMed PMID: 28158197]