Continuing Education Activity
Medullary thyroid cancer is a rare neuroendocrine tumor originating from the thyroid parafollicular C cells, which produce the hormone calcitonin. This malignancy represents 1% to 5% of all thyroid cancers and can occur sporadically or as part of hereditary syndromes, such as multiple endocrine neoplasia (MEN) types 2A and 2B or familial medullary thyroid cancer (FMTC). However, medullary thyroid cancer accounts for approximately 13% of thyroid cancer-related mortality, indicating a disproportionate impact on outcomes.
Medullary thyroid cancer typically presents as a thyroid nodule but may also manifest as a mass due to metastatic disease or symptoms related to elevated calcitonin levels, such as diarrhea or flushing. Diagnosis of this neoplasm typically starts with identifying a thyroid nodule, followed by ultrasound, fine needle aspiration biopsy, and measurement of tumor biomarkers like calcitonin. Genetic testing is crucial for identifying mutations present in all hereditary cases and some sporadic cases.
The poor prognosis associated with medullary thyroid cancer is partly due to delayed diagnosis and its higher propensity for metastasis compared to differentiated thyroid carcinoma, making early and accurate staging critical. Treatment primarily involves total thyroidectomy with lymph node dissection. This activity for healthcare professionals is designed to enhance the learner's competence in recognizing the clinical features of medullary thyroid cancer, performing the recommended evaluation and staging, and implementing an appropriate interprofessional management approach to improve patient outcomes.
Objectives:
Identify the underlying etiologies of medullary thyroid cancer.
Implement evidence-based guidelines for the surgical management of medullary thyroid cancer.
Screen individuals for high-risk factors of medullary thyroid cancer.
Apply interprofessional team strategies to improve care coordination and outcomes for patients affected by medullary thyroid cancer.
Introduction
Medullary thyroid cancer (MTC) is a neuroendocrine tumor that accounts for approximately 1% and 5% of all thyroid cancer cases.[1][2] These tumors originate from the parafollicular C cells of the thyroid gland, which are responsible for producing calcitonin. MTC presents in a sporadic form, comprising about 75% of cases; it can also be associated with hereditary syndromes, such as multiple endocrine neoplasia (MEN) types 2A and 2B and familial MTC (FMTC).[3] MEN2A typically features MTC, primary hyperparathyroidism, and pheochromocytoma, representing 95% of MEN2 cases.
Patients with a known genetic predisposition may benefit from early diagnosis through screening. Sporadic MTC cases often present similarly to other thyroid cancers, most commonly with a thyroid nodule. Therefore, the diagnostic pathway of MTC is the same as that of other thyroid cancers. Due to its tendency for metastatic spread, clinical lymphadenopathy and distant metastatic disease are more common in MTC. MTC may also be associated with symptoms related to elevated calcitonin levels (eg, diarrhea or flushing), ectopic Cushing syndrome, or positive familial screening.
Treatment generally involves total thyroidectomy with cervical lymph node dissection. Advances in the understanding of the molecular pathogenesis of MTC have led to the approval of targeted agents for use in certain patients with advanced disease. Since MTC arises from parafollicular C cells, it does not concentrate iodine, making radioactive iodine uptake studies and ablation ineffective as treatment options.[4]
Etiology
MTC arises de novo in approximately 75% of patients. About half of these sporadic tumors are associated with somatic RET mutations, while the remaining cases are linked to mutations in the RAS pathway. The underlying drivers for these mutations remain unknown. The remaining 25% of MTC cases are associated with a genetic predisposition, most commonly MEN 2A, followed by MEN 2B and FMTC.[5]
All patients with inherited forms of MTC carry germline mutations in the ret proto-oncogene that produces a receptor tyrosine kinase that plays a crucial role in several critical cellular pathways. Mutations in a specific area of the gene disrupt certain chemical bonds, causing RET proteins to pair up incorrectly. This results in uncontrolled activation of the pathways (eg, phospholipase Cγ/protein kinase C, c-Jun N-terminal kinases, proto-oncogene Src-related kinases phosphatidyl-inositol-3-kinase, and mitogen-associated protein kinase) leading to increased cell growth and changes in cell differentiation and migration.[3][6] Please see StatPearls' companion reference, "Multiple Endocrine Neoplasia Type 2," for more discussion of this syndrome.
Epidemiology
MTC accounts for approximately 1% to 5% of all thyroid cancers in the United States and globally.[1][2] Sporadic MTC typically peaks in incidence during the fifth or sixth decade of life. In contrast, MTC associated with MEN2A or 2B can present as early as childhood or the first decade of life, while patients with FMTC usually present in the second or third decade. The age of presentation is closely linked to specific RET gene mutations. Unlike other types of thyroid cancer, MTC does not exhibit a female predominance.[4]
Pathophysiology
MTC arises from parafollicular C cells in the thyroid gland, originating from the neural crest. RET mutations, associated with all hereditary and 50% of sporadic tumors, involve alterations in the ret proto-oncogene, which encodes tyrosine kinase receptors, including epidermal growth-factor receptors (EGFR) and vascular endothelial growth-factor receptors (VEGFR). Other pathways implicated in MTC include include the CTNNB1, RB, and mTOR pathways. Sporadic tumors that lack RET mutations often have mutations in the RAS pathway.[7] These molecular alterations ultimately lead to C-cell hyperplasia and, eventually, carcinoma. As parafollicular C cells produce calcitonin, calcitonin is a valuable marker for monitoring tumor growth and recurrence. However, this is not specific enough for diagnosis, as calcitonin can be falsely elevated in other conditions.[3] Patients with advanced MTC often suffer from diarrhea and flushing due to the tumor-secreting calcitonin and, at times, other peptides like an adrenocorticotropic hormone (ACTH) and calcitonin gene-related peptide. In rare instances, the tumor's ACTH production may lead to Cushing syndrome.[8][9]
Histopathology
Gross Pathology
MTC typically presents as a white, tan, or red mass. MTC is usually a single, unifocal, well-circumscribed mass in its sporadic form. In contrast, familial MTC is often bilateral and multifocal. Tumors commonly occur in the upper third of the thyroid gland, where the density of parafollicular cells is highest.[5]
Microscopic Appearance
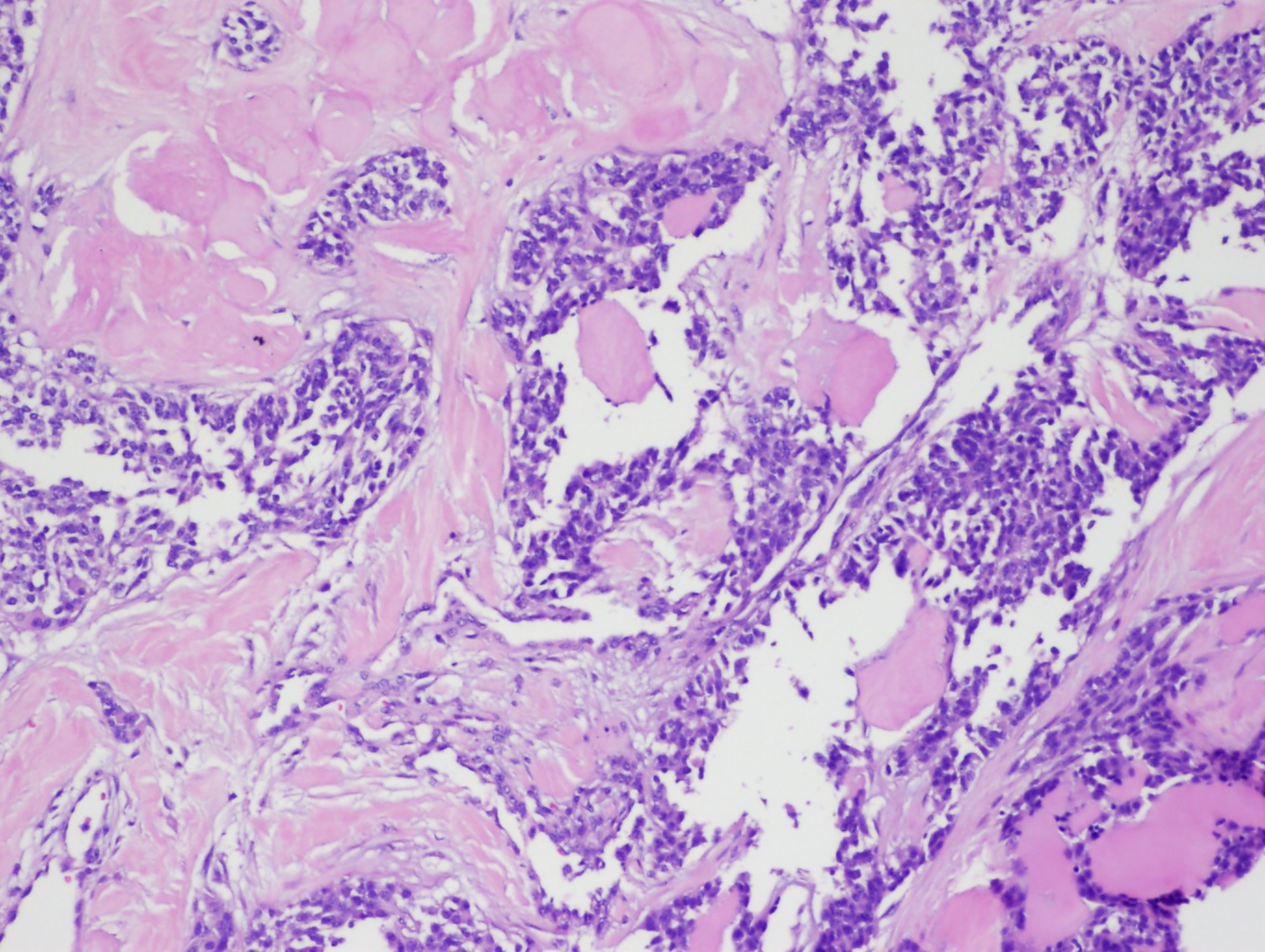
Classic cytological features seen in fine-needle aspiration (FNA) specimens of MTC include a scattered arrangement of polygonal or triangular cells with azurophilic granules and eccentrically located nuclei displaying coarse, granular chromatin (see Image. Medullary Thyroid Carcinoma). Tumors may exhibit amyloid deposits derived from calcitonin as well as coarse calcifications. Variants of MTC are common, and high-grade features such as tumor necrosis, increased mitotic activity, and a high Ki-67 proliferation index are associated with poorer outcomes.[5]
History and Physical
Clinical History
Patients with MTC typically present with a thyroid nodule between 40 and 60 years of age. The clinical history should focus on symptoms caused by local compression or invasion, such as hoarseness, dysphagia, or dyspnea. Additionally, clinicians should attempt to determine the lesional growth rate based on the timing of clinical features. Obtaining a detailed family history is crucial if a familial cause is suspected. Patients with hereditary syndromes may present with thyroid nodules or signs of metastatic disease or may be identified as having MTC through close screening. Symptoms of more distant metastatic disease may include diarrhea and flushing due to the tumor's secretion of calcitonin and other active peptides, including ACTH and calcitonin gene-related peptide.[4][5]
Physical Examination
Identifying the primary lesion and its association with surrounding structures is essential during the physical examination. Approximately 15% to 50% of patients have clinically evident cervical lymphadenopathy at diagnosis. Therefore, a careful lymph node examination should be performed.[10]
Evaluation
Since MTC often presents as a thyroid nodule, the diagnostic approach parallels evaluating any thyroid nodule. Evaluation includes performing a thyroid ultrasound and obtaining an FNA biopsy of the lesion. Genetic testing should be performed in all cases of MTC to identify germline mutations and other targetable mutations. In patients with hereditary MTC, identifying RET mutations and the specific codon affected is crucial for determining prognosis. Screening for pheochromocytoma and primary hyperparathyroidism is recommended if there is a positive history of MEN syndrome or if genetic testing results are pending.[11]
Baseline serum calcitonin and carcinoembryonic antigen (CEA) measurements are essential tumor markers in MTC. A cervical ultrasound should also be performed to assess for lymphadenopathy. For patients with calcitonin levels greater than 500 ng/mL, extensive disease on neck ultrasound, or clinical or radiologic evidence of distant metastasis, additional imaging is necessary for comprehensive staging. Options include contrast-enhanced computed tomography (CT) of the neck and chest, liver imaging via CT or magnetic resonance imaging (MRI), and positron emission tomography (PET)/CT, with gallium Ga 68 dotatate PET (Ga-68 PET/CT)demonstrating superior sensitivity compared to fludeoxyglucose-18 PET (FDG-PET).[5] Additionally, a laryngeal examination is advised for patients with voice changes, a history of prior surgery involving the recurrent laryngeal or vagus nerves, or extensive neck disease.[5]
Treatment / Management
Management of MTC requires a multidisciplinary and individualized approach based on factors such as whether the disease is sporadic or hereditary and whether it is resectable or unresectable. In sporadic MTC, surgical intervention is the primary treatment, with total thyroidectomy and bilateral central neck dissection recommended for tumors ≥1 cm or with bilateral involvement. For smaller, unilateral tumors, total thyroidectomy is advised, and neck dissection may be considered based on specific clinical factors. Since MTC cells do not take up iodine, sodium iodide I 131 (131I or RAI) therapy is ineffective, requiring postoperative levothyroxine therapy to maintain normal thyroid-stimulating hormone (TSH) levels; TSH suppression is not indicated in MTC.[5][4]
In hereditary MTC, the decision to perform prophylactic thyroidectomy is guided by mutation analysis and calcitonin levels, with early surgical intervention recommended for patients harboring high-risk RET mutations. For cases of recurrent or persistent disease without distant metastasis, further imaging and possible surgical resection are warranted.[5][4]
For unresectable MTC, systemic therapy options include tyrosine kinase inhibitors (TKIs), including vandetanib and cabozantinib, as well as RET-specific inhibitors, such as selpercatinib and pralsetinib. In patients with specific tumor characteristics that have progressed after prior therapies, pembrolizumab may also be considered. Regular monitoring and a collaborative, multidisciplinary approach are critical to optimize outcomes in MTC management.[5][4]
Differential Diagnosis
Patients with medullary thyroid cancer have a clinical presentation similar to other thyroid diseases. The following differential diagnoses must be kept in mind while evaluating these patients.
- Papillary thyroid cancer
- Follicular thyroid carcinoma
- Oncocytic (Hürthle) thyroid carcinoma
- Anaplastic thyroid carcinoma
- Intestinal carcinoid tumor
- Thyroid lymphoma
- Thyrotoxicosis
- Toxic nodular goiter
- Graves disease
Surgical Oncology
Sporadic Disease
Surgical intervention is the cornerstone of treatment for MTC. For patients presenting with tumors larger than 1 cm or with bilateral thyroid involvement, total thyroidectomy with bilateral central neck dissection (level 6) is recommended. For tumors smaller than 1 cm and unilateral disease, total thyroidectomy alone may be sufficient. Central neck dissection may be performed on an individualized basis if clinically involved cervical lymph node basins should be performed in patients with proven lymph node metastases.[4]
Hereditary Disease
Patients with germline RET mutations should undergo prophylactic thyroidectomy, with decision-making tailored based on mutation analysis and calcitonin levels. Specialized teams at tertiary care centers should conduct management. Patients with high-risk RET mutations are candidates for early thyroidectomy, particularly those with MEN2A, where surgery is generally recommended before patients reach 5 years of age. High-risk mutations include codons 609 and 611, among others, though specific mutation details are beyond this scope. For MEN2B, associated with an earlier onset, high-risk mutations necessitate thyroidectomy by age 1 year. Patients with low-risk RET mutations may be monitored with regular ultrasound and serum calcitonin surveillance, with surgery considered upon signs of MTC development.[4]
In patients with MEN2A, central neck dissection is only indicated for patients with ultrasound evidence or direct visualization of lymph node metastases at surgery or those with calcitonin levels exceeding 40 pg/mL. For cases associated with concomitant hyperparathyroidism, the surgeon should preserve or autotransplant an amount of tissue equivalent to 1 healthy parathyroid gland. A bilateral central neck dissection should be considered for all patients with MEN2B.[4]
Recurrent and Persistent Disease
In cases of biopsy-confirmed recurrent disease without distant metastasis, surgical resection is recommended.[4]
Surveillance
Postoperative assessment for residual disease includes serum calcitonin and CEA measurements at 3 months. If values are within normal ranges, repeat testing is recommended every 6 to 12 months. For elevated serum levels, further imaging is indicated. A neck ultrasound is performed for calcitonin levels <150 pg/mL. For levels >150 pg/mL, additional imaging (contrast-enhanced CT or MRI of the chest and abdomen, a bone scan, or spinal MRI) is recommended alongside the neck ultrasound. Brain MRI is reserved for patients with neurological symptoms.[4]
For asymptomatic patients with elevated tumor markers and persistently negative imaging, disease status should be monitored closely. In patients with residual thyroid tissue from incomplete initial surgery, a reoperation should be performed if feasible.[5][4]
Radiation Oncology
External beam radiation therapy has not been extensively studied as an adjuvant treatment for MTC. External beam radiation therapy for MTC can be considered for patients with grossly incomplete resections when further surgery is not an option. External beam radiation therapy can also be used to alleviate pain from bone metastases. Evidence regarding the optimal treatment volumes for MTC is limited, but intensity-modulated radiation therapy is preferred.[5][4]
Medical Oncology
Systemic Therapy
Tyrosine Kinase Inhibitors
For patients with unresectable and symptomatic MTC, TKIs such as the following agents may be indicated:
- Vandetanib: This TKI is an oral receptor kinase inhibitor targeting RET, EGFR, and VEGFR. In a phase 3 study involving 331 patients with advanced, unresectable, or metastatic MTC, vandetanib demonstrated improved progression-free survival (PFS) compared to placebo.
- Cabozantinib: This medication is also an oral multikinase inhibitor that targets MET, RET, and VEGFR2. The phase 3 EXAM study demonstrated improved PFS in patients with advanced or metastatic MTC treated with cabozantinib.[12][13]
RET-Specific Inhibitors
For patients with ret-mutant metastatic or nonresectable MTC, RET-specific inhibitors, including the following, are the preferred treatment options:
- Selpercatinib: Selpercatinib was evaluated in the phase 3 randomized LIBRETTO-531 trial, where it was compared to cabozantinib or vandetanib as first-line treatment for progressive ret-mutant MTC. At the 12-month follow-up, selpercatinib demonstrated a median PFS that was not yet reached, compared to 16.8 months for the comparator group.
- Pralsetinib: This RET-specific inhibitor was assessed in the phase 1 to 2 ARROW study, which included 92 patients with ret-mutant MTC. The overall response rate was 60% in patients who had received prior vandetanib and cabozantinib and 74% in patients with no previous treatment with these agents.[14][15][16]
PD-1 Inhibitors
Pembrolizumab may be an option for patients with TMB-H (tumor mutational burden-high, ≥10 mut/Mb) disease or those with microsatellite instability-high or mismatch repair-deficient tumors that have progressed following previous treatments, particularly when no satisfactory alternatives exist. Data from the KEYNOTE-158 study support this indication.[17]
Staging
The eighth edition of the American Joint Commission on Cancer (AJCC) Cancer Staging Manual outlines the tumor-node-metastasis (TNM) system for MTC. However, this classification omits several key prognostic factors, including the patient's age at diagnosis. Specifically, patients aged 40 and older may have different outcomes not reflected in the TNM staging.[4] The following criteria is used for TNM staging:
- Tumor (T)
- TX: Primary tumor cannot be assessed
- T0: No evidence of primary tumor
- T1: Tumor ≤2 cm in greatest dimension limited to the thyroid
- T1a: Tumor ≤1 cm in greatest dimension limited to the thyroid
- T1b: umor >1 cm but ≤2 cm in greatest dimension limited to the thyroid
- T2: Tumor >2 cm but ≤4 cm in greatest dimension limited to the thyroid
- T3: Tumor ≥4 cm or with extrathyroidal extension
- T3a: Tumor ≥4 cm in greatest dimension limited to the thyroid
- T3b: Tumor of any size with gross extrathyroidal extension invading only strap muscles (sternohyoid, sternothyroid, thyrohyoid, or omohyoid muscles)
- T4: Advanced disease
- T4a: Moderately advanced disease; tumor of any size with gross extrathyroidal extension into the nearby tissues of the neck, including subcutaneous soft tissue, larynx, trachea, esophagus, or recurrent laryngeal nerve
- T4b: Very advanced disease; tumor of any size with extension toward the spine or into nearby large blood vessels, gross extrathyroidal extension invading the prevertebral fascia, or encasing the carotid artery or mediastinal vessels [4]
- Nodes (N)
- NX: Regional lymph nodes cannot be assessed
- N0: No evidence of locoregional lymph node metastasis
- N0a: ≥1 cytologically or histologically confirmed benign lymph nodes
- N0b: No radiologic or clinical evidence of locoregional lymph node metastasis
- N1: Metastasis to regional nodes
- N1a: Metastasis to level VI or VII (pretracheal, paratracheal, or prelaryngeal/Delphian, or upper mediastinal) lymph nodes. This can be unilateral or bilateral disease
- N1b: Metastasis to unilateral, bilateral, or contralateral lateral neck lymph nodes (levels I, II, III, IV, or V) or retropharyngeal lymph nodes [4]
- Metastasis (M)
- M0: No distant metastasis
- M1: Distant metastasis present [4]
American Joint Commission on Cancer Prognostic Stage Classification
Based on the TMN assessment, the tumor may be staged according to the following AJCC staging classification:
- Stage I: T1, N0, and M0
- Stage II: T2, N0, and M0 or T3, N0, and M0
- Stage III: T1 to T3, N1a, and M0
- Stage IVA: T4a, any N, and M0 or T1 to T3, N1b, and M0
- Stage IVB: T4b, any N M0
- Stage IVC: Any T, any N, and M1 [4]
Prognosis
The prognosis depends on the patient's age, histologic grade, and surgical resection status. Older patients, patients with higher-grade lesions, and those with incomplete surgical resection have a worse prognosis. The 5-year survival rate for stages I to III is 93% compared to 28% for stage IV.
Other risk factors include patient age, nodal involvement, vascular invasion, and elevated calcitonin levels. Additionally, histological features such as mitotic rate and tumor necrosis have been linked to disease-specific outcomes. An international grading system has been developed to enhance prognostic predictability, categorizing MTC into low-grade and high-grade classifications. High-grade MTC is associated with a higher risk of local recurrence, distant metastasis, and poorer overall survival compared to low-grade tumors.[18][19]
Complications
As MTC advances, it can lead to various complications. Local spread of the tumor may cause issues, including difficulty swallowing or breathing. Additionally, the cancer can metastasize to lymph nodes, lungs, liver, or bones, resulting in pain and potential organ dysfunction. Elevated calcitonin levels can cause symptoms like flushing and diarrhea.
Additionally, surgical intervention (eg, total thyroidectomy) may damage nearby structures, including the recurrent laryngeal nerve, leading to voice changes or difficulty swallowing. Patients may also experience hypoparathyroidism, requiring calcium and vitamin D supplementation. Targeted therapy with TKIs can cause adverse effects, including hypertension, diarrhea, and fatigue. Patients may also experience skin rashes and changes in liver function. RET-specific inhibitors can lead to adverse effects, including elevated liver enzymes, gastrointestinal issues, and fatigue. Pembrolizumab, an immune checkpoint inhibitor, may cause immune-related complications, eg, colitis, endocrine disorders, pneumonitis, and skin reactions.
Deterrence and Patient Education
MTC is an aggressive tumor with a poor prognosis when diagnosed at advanced stages. It often appears as a thyroid nodule, but the initial diagnosis may involve metastatic disease in up to 50% of cases. Patients should be informed about the hereditary forms of MTC, and genetic counseling is essential. Risk assessment is based on specific RET mutations, which should guide recommendations for risk-reducing thyroidectomy.
After a total thyroidectomy, patients need education about lifelong thyroxine treatment. Additionally, patients should have physical examinations every 6 months for the first 2 years and annually thereafter. Serum calcitonin and CEA levels should be monitored, initially 2 to 3 months after surgery, then twice a year for 2 years, and then annually to detect recurrence or distant metastasis. If calcitonin levels are elevated, neck ultrasounds and further imaging tests, eg, CT, MRI, or PET/CT, should be considered.
Enhancing Healthcare Team Outcomes
Enhancing healthcare team outcomes for patients with MTC relies on collaborative efforts from physicians, advanced practitioners, nurses, genetic counselors, pharmacists, and other health professionals. Effective interprofessional communication and care coordination are essential to align all team members in delivering patient-centered care. Clinical expertise, empathy, and ethical decision-making are critical in managing hereditary syndromes like MEN and familial MTC (FMTC).
Each team member plays a unique role. Physicians lead diagnostics and management, while nurses offer critical symptom management and patient education. Genetic counselors identify at-risk family members and guide appropriate testing, while pharmacists manage medications and monitor potential drug interactions, especially with targeted therapies such as tyrosine kinase inhibitors. Fostering a culture of teamwork and shared responsibility enhances patient safety, improves outcomes, and optimizes overall team performance in MTC management.