Introduction
Immunoglobulin E is 1 of the 5 classes of immunoglobulins (IgM, IgG, IgD, IgA, IgE). IgE has a unique chemical structure and various physiological functions, such as type I hypersensitivity reactions, parasitic infections, autoimmune processes, and venom protection.[1] It was the last of the immunoglobulin family to be discovered. Since then, it has spurred vast research to characterize its biochemistry and role in disease processes. Technological advancements have also been used to evaluate patient IgE levels and new pharmacological therapies that may inhibit its function altogether.[2] The following article aims to provide an overview detailing the biochemical structure and function of IgE, as well as some examples of its clinical implications.
Molecular Level
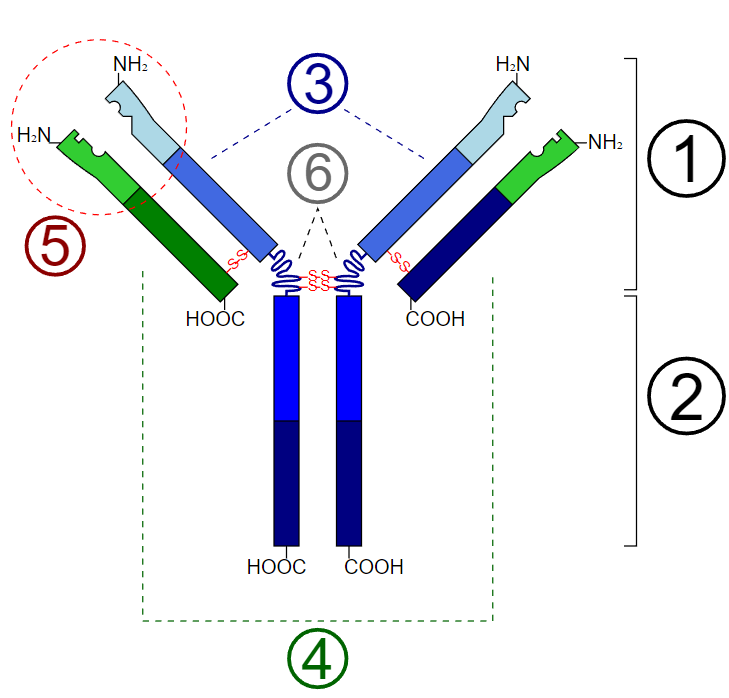
All immunoglobulin molecules have a common structural framework characterized by 2 identical small polypeptide chains (light chains) and 2 identical large polypeptide chains (heavy chains). The fragment crystallizable (Fc) region of the antibody is comprised solely of the 2 heavy chains and is responsible for binding to cellular receptors. The fragment antigen-binding (Fab) region of the antibody is where antigen recognition and binding occur and is made up of 2 heavy chains continuous with the Fc region and 2 light chains. These chains also have variable and constant regions. At the antigen binding sites, both polypeptide chains contain variable regions whose amino acid sequences can be altered for affinity maturation to occur for a given antigen. The variable heavy (VH) and variabl light (VL) chains are organized into layers of antiparallel B-pleated sheets, allowing the antigen to bind between these 3-dimensional immunoglobulin folds. The rest of the polypeptide regions are known as constant regions, and they are held together by strong dipeptide bonds to stabilize the 2 chains.[3]
Some antibodies, like IgM, can have 5 duplicates of the structure above to form a pentamer capable of binding up to ten antigens. However, IgE is a monomer composed of 2 epsilon-heavy chains and 2 light chains, thus capable of binding a total of 2 antigens, which occurs through the variable regions of the light and heavy chains that create unique antigen-specific binding sites. The C-terminal regions of the heavy chains are composed of 4 C-epsilon dimers, C-epsilon 1-4.[4] The chemical structure of these dimers is essential for binding IgE-specific cellular receptors Fc-epsilon RI and CD23, which will be discussed in more detail later. Some immunoglobulins, such as IgG, contain a "hinge" region near the molecule's center. IgE is unique because it lacks this hinge region and is replaced by the C-epsilon2 domain. There is a theory that the lack of this hinge region allows the IgE molecule to adopt a more flexible conformation while interacting with its receptors. Furthermore, the IgE molecule is more heavily glycosylated than other antibodies, containing 7 N-linked glycosylations on each heavy chain, which is necessary for binding to a high-affinity cellular receptor, Fc-epsilon-RI (see Diagram. Basic Unit of Immunoglobulin [Antibody]).[4]
Function
The biochemical considerations mentioned determine the structure of IgE and drive the function of allowing IgE to bind to given antigens and IgE-specific cellular receptors. For IgE to accomplish its function, the Fc portion of the antibody must bind to a given cellular receptor located on a particular type of cell, such as a mast cell, eosinophil, etc. Although many cellular receptors have been identified and studied, the main ones are Fc-epsilon-RI and Fc-epsilon-RII, or CD23. [5] Fc-epsilon-RI, or the high-affinity receptor, is mainly located on mast cells, basophils, dendritic cells, macrophages, and eosinophils. It is thus responsible for immediate hypersensitivity reactions, parasitic immunity, enhanced cytokine production, and antigen presentation. The other receptor, CD23, or the low-affinity receptor, is primarily expressed on B-cells, T-cells, and antigen-presenting cells (APCs) and is responsible for IgE production homeostasis, facilitated antigen presentation (FAP), as well as transportation of IgE across airway and gut epithelia.[6]
Pathophysiology
The role of IgE is central in allergy sensitization and atopic disorders such as allergic rhinitis, asthma, and atopic dermatitis. These disorders manifest due to type I hypersensitivity reactions involving IgE and other immune cells to produce the clinical symptoms seen in those disorders ultimately. It starts with an initial exposure to an antigen or allergen, which is taken up and processed by a dendritic cell or macrophage, which presents the antigen to a T-cell. In the presence of cytokine mediators IL-4 and IL-13, these T-cells are induced to differentiate into TH2 helper T-cells capable of presenting the antigen to B-cells.[6] The B-cells then undergo class-switching recombination events to produce Immunoglobulin E antibodies capable of binding to the presented antigen at the "antigen binding site" of the Fab portion of the antibody. Once this initial sensitization to antigen has occurred, more immunological events can ensure a more robust IgE response. The CD23 receptor on intestinal or airway epithelial cells can transport antigen-IgE complexes across the epithelium to bind Fc-epsilon-RI receptors on mast cells, macrophages, and other immune cells to promote inflammation, cytokine production, and more local IgE production. Furthermore, via interaction with the CD23 receptor on B-cells and myeloid cells, a process known as FAP can occur.
FAP involves antigen-IgE complexes binding to CD23 receptors on B-cells, which then present the peptides to T-cells to facilitate further IgE antibody production. [7] This approach contrasts with the classical antigen presentation pathway described above. Instead of T-cells being the primary antigen presenters to the B-cell, it is now antigen-IgE complexes formed in the periphery, which may or may not be related to the initial antigen. This has clinical importance because, via facilitated antigen presentation, the B-cells will ultimately present a diverse array of peptides to T-cells to clear it for further immunoglobulin production. In conclusion, this is a mechanism for epitope spreading, where an antibody response for 1 antigen can cause the production of antibodies to different antigens.[6] This could be why individuals can develop allergies to multiple antigens resulting in a phenomenon known as "atopic march."[8] For example, a child could have atopic dermatitis and later in life develop asthma. In conclusion, facilitated antigen presentation produces a more robust immune response to a given antigenic exposure that could be responsible for sensitization to multiple unrelated antigens.
Once initial exposure to an antigen has occurred, and an immune response has been activated, antigen-specific IgE will remain bound via the Fc-epsilon RI receptor to mast cells and basophils. It has been shown that binding alone of IgE to that receptor cause IgE-dependent upregulation of mast cell Fc-epsilon RI receptor expression, which allows these cells to bind more IgE antibodies.[6] Furthermore, it is essential to note that IgE has a short half-life in plasma, usually less than a day. However, IgE bound to a high-affinity receptor (Fc-epsilon RI) can remain attached to mast cells in tissues for weeks to months.[4] Later, upon re-exposure to a given antigen, crosslinking of adjacent Fc-epsilon RI-bound IgE antibodies occurs and binds the antigen, which causes subsequent aggregation of Fc-epsilon RI receptors. Once this happens and there is a sufficient aggregation of these receptors with bound antigen, it causes mast cells and basophils to release preformed chemical mediators stored in cytoplasmic granules such as histamine, serotonin, tryptase, prostaglandins, leukotrienes, and eosinophil or neutrophil chemotactic factors.[6]
Clinical Significance
It is essential to note that not everyone exposed to environmental antigens will generate a strong immune reaction described above. Certain people, known as "atopics," have a genetic predisposition for developing type I hypersensitivity responses to antigens in the environment. Clinically, the manifestations of this type one hypersensitivity reaction are usually due to the actions of the released chemical mediators such as histamine, leukotrienes, and other cytokines. Histamine release causes increased vascular permeability, smooth muscle constriction, and mucus secretion. Leukotrienes B4, C4, D4, and E4 are also often secreted and responsible for chemotaxis, inflammation, and anaphylaxis. Furthermore, if in enough quantity, histamine and other mediators can cause systemic vascular effects such as vasodilation and increased vessel permeability, causing hypotension and edema.[6] This could lead to anaphylactic shock and death if severe and poorly managed.
The manifestations can occur in many different areas of the body. Involvement of the upper airway can cause many patients to suffer from mild seasonal allergies or allergic rhinitis. Due to repeated inhalation exposure to pollen and other outdoor antigens, type I hypersensitivity occurs, and mast cell degranulation causes epiphora, rhinorrhea, cough, and other symptoms. Additionally, the involvement of the lower airway in patients with atopic asthma experiences an immune response to aeroallergens resulting in airway smooth muscle constriction, increased mucus production and inflammation that manifests as obstructive lung disease. The skin can often be involved, and due to vasodilation and dermal edema, urticaria or "wheals’"can be evident.[9]
Another skin disease that has been studied concerning the roles of IgE and hypersensitivity is atopic dermatitis or eczema. Although eczema is a multifactorial disease involving genetics, barrier dysfunction, and immune dysregulation, some evidence suggests IgE-mediated hypersensitivity may also play a role.[10] The inflammatory infiltrates of AE lesions are composed mainly of CD4+ helper T-cells, with a predominance of TH2 cells, which are responsible for initiating IgE class switching by B-cells. Certain aeroallergens were found to penetrate compromised skin in eczema patients and interact with Langerhans cells. Furthermore, these same cells were discovered to have IgE binding structures on their surface.[11] A correlation has also revealed that up to 80% to 85% of patients with atopic dermatitis have elevated IgE levels.[12] One study even conducted atopy patch tests on eczema patients and correlated their allergen-specific serum IgE. It showed that most patients (40%, N=151) with positive atopy patch test for D. pteronyssinus (house dust allergen) also had elevated serum IgE specific for that antigen. This study also demonstrated that there were patients (9%) with a positive atopy patch test and no elevation in serum IgE, in addition to other patients with a negative patch test.[11] Thus, given the known multifactorial nature of atopic dermatitis, it is likely that some correlation may exist between IgE-mediated hypersensitivity and eczema. However, the exact nature of this relationship requires further investigation.
IgE has also been implicated in the defense against parasitic organisms such as helminths. Helminthic infections are usually the result of soil transmission and infect up to one-third of people worldwide.[13] When the immune system encounters this type of parasite, the B-cell class switch to IgE antibodies, which then "coat" the parasite via binding many antibodies. Following this, effector cells such as eosinophils and mast cells recognize Immunoglobulin E bound to the helminth and can then attach the Fc portion of the antibody via the Fc-epsilon RI receptor. Once connected, subsequent reactions occur, such as further cytokine release and histamine production by mast cells, as well as major essential protein and eosinophil peroxidase production by eosinophils.[14] These substances are toxic to the helminth and can thus result in parasitic killing and the ultimate clearance of the invader.
As depicted in this article, Immunoglobulin E has a specific biochemical structure and function that guides its unique role in disease processes such as type I hypersensitivity reactions, helminthic infections, and more. The research in the past 50 years regarding IgE has established a better idea of its role in disease. Not only has this aided in a more in-depth clinical understanding of IgE-related health disorders, but it has even spurred novel treatment modalities. A humanized monoclonal antibody named omalizumab has been engineered, which targets the IgE antibody, thus rendering it ineffective in producing associated symptoms. This drug has been shown to provide benefits and relief in patients with moderate to severe asthma and intermittent and persistent allergic rhinitis.[15] Despite this progress, much more remains to be learned about the exact function of this immunoglobulin in those diseases in which some correlation has already been established, as well as novel clinical relationships yet to be discovered.