Continuing Education Activity
Carnitine deficiency is a condition characterized by low carnitine levels in the body. Carnitine deficiency could be primary (due to defect in carnitine transport) or secondary to other conditions. Primary carnitine deficiency (PCD) is inherited as an autosomal recessive disorder. The spectrum of presentation in PCD varies from being asymptomatic to sudden onset. To avoid the complications associated with this condition, PCD must be promptly diagnosed and treated with L-carnitine. Secondary carnitine deficiency (SCD) could result from multiple causes, either from a decrease in carnitine intake or more commonly from an increase in renal excretion as acylcarnitine. This activity reviews the causes, pathophysiology, and clinical presentation of conditions causing carnitine deficiency. It reviews the evaluation and treatment strategies of carnitine deficiency and highlights the role of the interprofessional team in evaluating and treating patients with this condition.
Objectives:
- Describe the etiology of carnitine deficiency.
- Outline the evaluation of carnitine deficiency.
- Review the management options available for carnitine deficiency.
- Outline interprofessional team strategies for improving care coordination and communication to manage carnitine deficiency conditions and improve outcomes.
Introduction
Carnitine (beta-hydroxy-gamma-trimethylammonium butyrate) is an indispensable water-soluble molecule derived from amino acids.[1][2] In non-vegetarians, dietary intake is the primary source of carnitine and accounts for almost three-fourths of the total body stores.[3] The main dietary source of carnitine is red meat, poultry, and dairy products.[4] The bioavailability of dietary carnitine is between 54% to 87%.[5] The remaining one-fourth of the carnitine pool can be produced endogenously from lysine and methionine mostly by the liver and kidneys.[3][6] Vegetarians have a relatively lower plasma carnitine level than non-vegetarians.[7] In strict vegetarians, most of the carnitine (>90%) is produced endogenously.[3] Despite variabilities in dietary carnitine intake, the plasma carnitine level is maintained within the normal range by an efficient renal reabsorption system while excessive carnitine is promptly excreted in the urine.[8] About 90% to 99% of the filtered carnitine is usually reabsorbed by the renal tubules.[3]
Carnitine plays a substantial physiological role in lipid metabolism and intermediary metabolic pathways.[5] Through the carnitine shuttle, carnitine helps in transporting the long-chain fatty acids from the cytoplasm to the mitochondrial matrix for subsequent degradation for beta-oxidation, which is detailed in the pathophysiology section (figure).[3] Plasma carnitine accounts for approximately 0.5% of the total body stores, and the remaining vast majority is found within the cells.[3][5] As carnitine is essential for fueling the exercising muscle through fatty acid oxidation and energy production via the Krebs’s cycle, more than 95% of total body carnitine is found in skeletal muscle.[5][7][9] The liver, heart, and kidneys have the rest of the carnitine stores.[5]
Etiology
Primary carnitine deficiency (PCD) due to carnitine transport defect is an autosomal recessive (AR) genetic disorder of the OCTN2 (organic carnitine transporter novel type 2) carnitine-transporter system encoded by the SLC22A5 gene.[4] Carnitine transport defect causes a significant carnitine depletion and decreases intracellular carnitine accumulation due to deficient active carnitine transport across the plasma membrane. PCD is characterized by low plasma carnitine levels, reduced intracellular carnitine, and increased urinary loss.[5] Carnitine is transported intracellularly via OCTN2, which is expressed predominantly in skeletal and cardiac muscles, and kidneys.[3] Decreased OCTN2 on the plasma membrane results in a reduced intracellular update of carnitine.[5] In kidneys, this results in reduced reabsorption of carnitine, and patients with PCD may lose up to 95% of the filtered carnitine in the urine.[6] The parents of a child with PCD, who are heterozygous carriers, may lose twice or thrice the levels of normal urinary excretion.[6] The plasma concentration of acyl-carnitine esters is also low in PCD.[5]
Epidemiology
The incidence of Primary carnitine deficiency is different based on ethnicity. The frequency of PCD varies between countries. In the U.S, the incidence is approximately 1: 142,000 based on newborn screening data.[10] In Japan, the incidence is approximately 1 in 40,000.[11] The Faroe Islands, an archipelago in the North Atlantic, report the highest incidence of PCD, which is 1 in 300.[12] Some patients with PCD have no or minimal symptoms, and this milder phenotype may remain undiagnosed throughout their entire life. Consequently, it is challenging to ascertain the exact prevalence of all PCD phenotypes.[13] The PCD distribution across the gender is expected to be equal as it is inherited in AR fashion. However, many women are diagnosed with PCD shortly after giving birth, following their infants being detected with low carnitine levels during newborn screening. Also, females may have more frequent clinical manifestations, especially given the stress of pregnancy.[14]
Pathophysiology
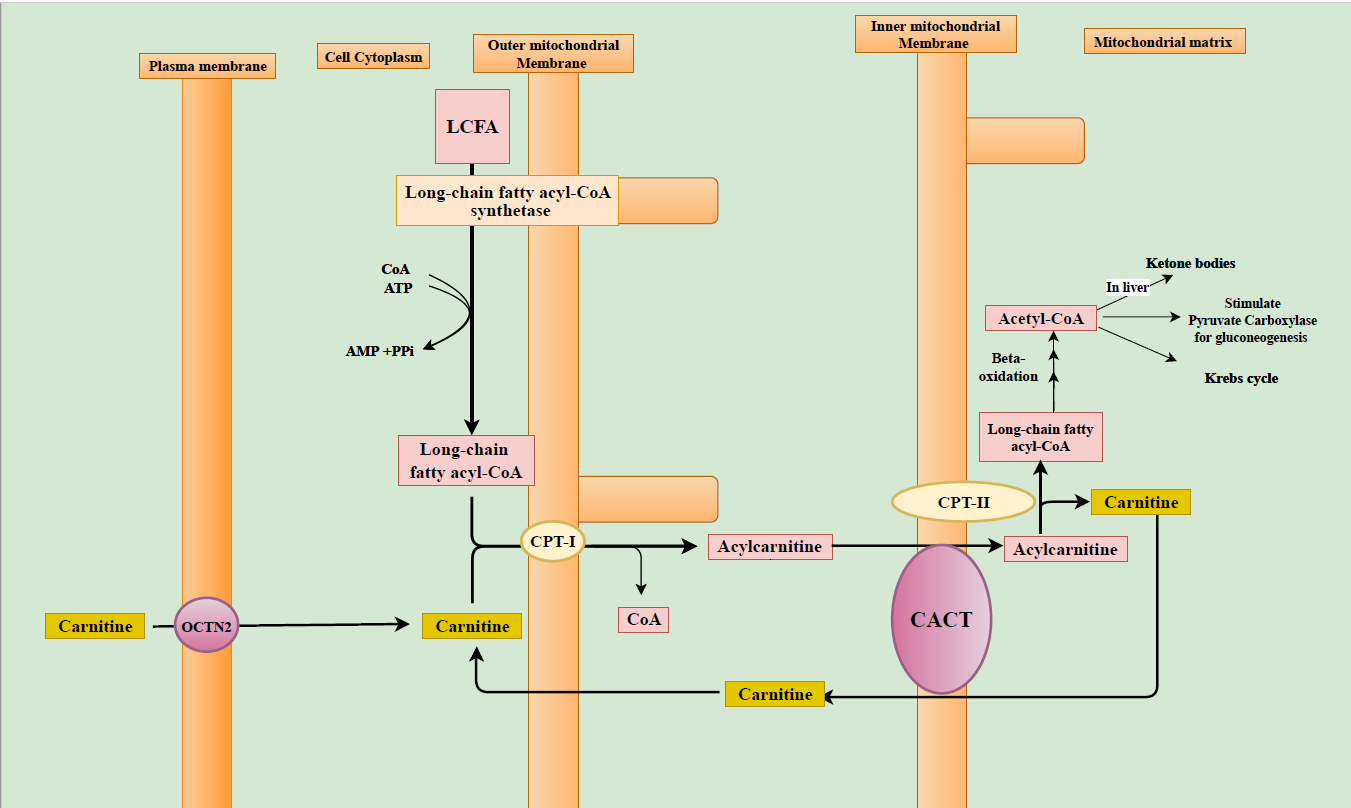
Under normal physiological conditions, fatty acids are the main source of energy during fasting.[6] Energy production from fatty acids occurs via beta-oxidation of fatty acids in the liver, heart, and skeletal muscles.[6] Beta oxidation of the long-chain fatty acid (LCFAs) occurs exclusively in the mitochondrial matrix. The mitochondrial membrane is impermeable to LCFAs and requires the obligatory carnitine shuttle (Figure). To be transported across the mitochondrial membrane, LCFAs are activated in the cytoplasm by conversion to long-chain fatty acyl-CoAs (LCFA-CoAs). This reaction is catalyzed by long-chain fatty acyl-CoA synthetase.[5] LCFA-CoAs then can diffuse through the outer mitochondrial membrane where they are converted to acylcarnitine in a presence of carnitine by the enzyme carnitine palmitoyltransferase-1 (CPT-I). In normal states, acylcarnitine formation reduces the proportion of acyl residues combined with coenzyme A (CoA) and improves the ratio between free CoA and acyl-CoAs.[6] Acylcarnitine crosses the inner mitochondrial membrane by the carnitine-acylcarnitine translocase (CACT) via the carnitine shuttle. In the mitochondrial matrix, acylcarnitine is converted back to LCFA-CoAs and free carnitine by the carnitine palmitoyltransferase-2 (CPT-II). When acylcarnitine crosses the inner mitochondrial membrane, simultaneously the free carnitine released by the CPT-II leaves the mitochondrial matrix by the action of CACT and this termed as the carnitine shuttle. Once they are inside the mitochondrial matrix, LCFA-CoAs are readily oxidized and results in acetyl-CoA production. Acetyl-CoA is subsequently used for energy and ketone bodies production. Acetyl-CoA is also an allosteric activator of pyruvate carboxylase that catalyzes the gluconeogenesis pathway which is active during catabolic states such as fasting.[6][15][16]
In carnitine deficient states, LCFAs cannot be effectively transported to the mitochondria matrix for oxidation and subsequent utilization in Kreb’s cycle and ketone body production. During periods of fasting, improper utilization of fatty acids impairs gluconeogenesis and characteristically leads to nonketotic or hypoketotic (no or minimal ketone body production respectively) hypoglycemia.[3] When fatty acid oxidation is impaired, glucose is readily consumed without replenishment from gluconeogenesis.[6] In carnitine deficiency states, various intermediary metabolic pathways such as Kreb's cycle, amino acid metabolism, and beta-oxidation of fatty acids are also affected. Fatty acids released from adipose tissues during fasting accumulate in various organs predisposing to their impaired function.[5][6] Accumulation of fat in the liver causes steatosis and impairment of ketone body production.[5][6][8] In the heart and skeletal muscles, this abnormal accumulation results in cardiomyopathy and myopathy respectively.[5][6] Heart derives two-thirds of its energy from free fatty acids and this predisposes patients with impaired carnitine metabolism to the development of cardiomyopathy.[6] Additionally, impaired lipid metabolism can affect the electrical rhythm of the heart resulting in arrhythmias.[6] The brain utilizes ketone bodies as an alternate energy source in fasting states. These ketones are derived from acetyl-CoA from fatty acid oxidation and in carnitine deficiency, this is defective.[6] Unstable energy and metabolic abnormalities can impair brain function with loss of consciousness and metabolic encephalopathy.[6][17]
History and Physical
The presentation of Primary carnitine deficiency is variable between patients in terms of severity.[3] The clinical manifestations and severity of symptoms of PCD can vary based on the age of onset and the involved organ systems. About half of the affected individuals present at a younger age, between three months and two years, with attacks of metabolic decompensation in a form of hepatic encephalopathy characterized by poor feeding, lethargy, and irritability. The attacks occur particularly during periods of catabolism when fatty acids oxidation and gluconeogenesis are mostly needed e.g. fasting or during common illnesses such as acute gastroenteritis.[3] These attacks are associated with hepatomegaly, nonketotic or hypoketotic hypoglycemia, lactic acidosis, hyperammonemia, and elevated hepatic enzymes (alanine aminotransferases and aspartate aminotransferases).[3]
Hyperammonemia occurs due to reduced expression of enzymes involved in the urea cycle and noted during acute metabolic decompensation.[6] The remaining half of the affected individuals present during childhood, between 2 and 4 years, with myopathic symptoms such as hypotonia, skeletal muscle weakness, exercise intolerance, episodes of rhabdomyolysis and myoglobinuria and elevated creatine kinase (CK).[3] Additionally, cardiac manifestations like dilated cardiomyopathy may occur and, if not promptly treated with carnitine may progress to heart failure and death.
Less frequently, adults with PCD may have no or mild symptoms (e.g. fatigue) but sudden cardiac death remains a risk in asymptomatic adults.[3][6] Some women have been diagnosed with PCD after their newborns were found to have low carnitine levels during neonatal screening.[3] Atypical manifestations such as anemia, respiratory distress, proximal muscle impairment, developmental regression, and arrhythmias have also been reported.[5][13]
Evaluation
When carnitine deficiency is suspected, the first step is to measure plasma carnitine levels.[13] Patients with PCD have low plasma free carnitine levels (< 5 μmol/L, normal 20–50 μmol/L).[4][6] Prompt referral to a metabolic specialist should be done and genetic testing is performed to confirm the diagnosis. Sequence analysis of the SLC22A5 gene, followed by the array comparative genomic hybridization (aCGH) if the sequence analysis is not conclusive. There is usually a poor correlation between genotype and phenotypic expression in PCD.[6][18] Some investigators have noted nonsense and frameshift mutations more commonly in symptomatic patients, and in asymptomatic mothers with PCD (identified by positive newborn screening of their infants), missense mutations and in-frame deletion are commonly reported.[18]
If genetic testing fails to confirm PCD diagnosis, functional assay such as the cultured skin fibroblast carnitine assay is the preferred test. In PCD, the OCTN2 transporter activity is <10% of normal controls.[14] Once PCD diagnosis is confirmed, the patient should undergo a series of investigations including echocardiogram, electrocardiogram, serum creatine kinase (CK), serum transaminases, and blood sugar levels.[14] Other biochemical tests such as plasma acylcarnitine profile and urine organic acid analysis may be required to evaluate other conditions resulting in secondary carnitine deficiency which are detailed in the differential diagnoses section.[13]
In the U.S., the newborn screening test includes evaluation for PCD. By tandem mass spectrometry method, low plasma free carnitine levels are detected in newborns with PCD. Due to placental transfer of carnitine from the mother to the fetus, low fetal plasma creatinine levels shortly after birth can reflect the maternal plasma carnitine levels. Some mothers are diagnosed following their infants being detected with carnitine deficiency in their newborn screening tests. Therefore, if the newborn screening detects low carnitine levels, both the baby and the mother are re-tested, after two weeks, to determine who has PCD.[14]
Treatment / Management
The mainstay of primary carnitine deficiency involves lifelong treatment with a high dose of oral L-carnitine (100 to 200 mg/kg daily dose in 3 divided doses).[3][4] The oral bioavailability of L-carnitine is 5% to 18%.[5] L-carnitine is a fairly safe medication, and few side effects associated with high doses include diarrhea and intestinal discomfort. Additionally, bacterial degradation of unabsorbed oral L-carnitine in the bowel results in trimethylamine, which has a peculiar fishy odor.[3][6] These side effects can be minimized by decrease the L-carnitine dose or treatment with a 7 to 10-day course of oral metronidazole, which could eradicate the intestinal bacterial overgrowth.[13]
Maintenance therapy with L-carnitine can improve plasma levels, and the dose is titrated based on the plasma levels and also on the response.[3] Continued L-carnitine administration prevents hypoglycemic episodes as well as cause improvement in myopathic symptoms. The muscle carnitine levels rise slightly (only up to 5% to 10% of controls), due to the abnormal OCTN2 which is unable to increase the uptake of carnitine into the myocyte adequately.[5][8] Carnitine enters mostly by passive diffusion from plasma and via low-affinity transporters, and this modest increase is enough to prevent muscle complications.[8]
Acute episodes of hypoglycemia in children with PCD are promptly treated with intravenous 10% dextrose and treatment of accompanying metabolic abnormalities (e.g., acid-base abnormalities), along with immediate carnitine supplementation.[8][17][8] It is crucial to avoid episodes of hypoglycemia in PCD by frequent feeding and avoiding fast states.[17]
Differential Diagnosis
Secondary carnitine deficiency (SCD) could result from multiple causes, either from a decrease in carnitine intake or more commonly from an increase in renal excretion. SCD may result from severe malnutrition, ketogenic diet, severe malabsorptive states, extremely preterm infants, and prolonged parenteral nutrition without adequate L-carnitine supplementation.[4][6] In conditions with renal tubular dysfunction (e.g., Fanconi syndrome), renal losses of carnitine are accentuated. Medications such as valproic acid, cyclosporine, pivampicillin, and some anti-cancer drugs (e.g., etoposide, vinblastine) can also contribute to carnitine deficiency.[6] Valproic acid therapy may deplete carnitine stores by various mechanisms such as increased urinary excretion as valproylcarnitine, decreased renal tubular reabsorption, and decreased endogenous production.[19] Similarly, pivampicillin antibiotic usage may cause reduce carnitine levels due to the formation of pivaloyl-carnitine ester, which is excreted in the urine.[5] The severity of SCD is generally less when compared to PCD as the plasma carnitine levels are relatively higher and hence easier to manage.[3][8]Patients with SCD, in general, do not have hepatic or cardiac involvement. However, they may have a moderate degree of skeletal muscular dysfunction.[20]
Similarly to PCD, hypoketotic hypoglycemia is highly suggestive for mitochondrial fatty acid oxidation (FAO) disorders (e.g., very long-chain acyl-CoA dehydrogenase or VLCAD, medium-chain acyl-CoA dehydrogenase or MCAD, etc.), or disorders of a defective mitochondrial carnitine-acylcarnitine cycle or simply referred to as disorders of carnitine shuttle (due to defects in enzymes, CPT-I, or CPT-II or CACT).[6][15] In most of the aforementioned SCD disorders with impaired fatty acid oxidation, the accumulation of acylcarnitine esters occurs.[5] These excessive acylcarnitines can inhibit carnitine reabsorption in the kidneys and are subsequently excreted in the urine resulting in SCD.[6][5] The accumulating acyl-carnitine in cardiac tissue can also induce damage.[6]
The clinical manifestations of FAO defects and carnitine shuttle disorders are heterogeneous and can be vague.[15] Similar to PCD, the most common manifestations include hypoketotic hypoglycemia, myopathies of both skeletal and cardiac muscles.[3][15][21][22] Most often, the manifestations are precipitated or worsened by either fasting or intercurrent illnesses.[15] The resulting metabolic abnormalities can be evaluated by the plasma acylcarnitine profile.[5][15] Diagnostic confirmation is by molecular testing methods and/or functional assays of the respective deficient enzyme.[15] Management includes prevention of fasting, frequent feeding, and providing nocturnal corn starch.[3][21] Diet therapy should focus on high carbohydrates and medium-chain triglycerides (which do not require carnitine shuttle) and low in long-chain fatty acids.[21] Management with L-carnitine is controversial in FAO disorders.[23]
Carnitine is also important in binding acyl residues derived from the intermediary metabolism of amino acids.[6] In disorders of organic acidemias (e.g., isovaleric acidemias, propionic acidemias, methylmalonic acidemias), SCD results from binding of the excessive acyl-CoA intermediate products.[6] It is also noteworthy to mention here that the defects in endogenous carnitine biosynthesis usually do not lower plasma carnitine levels as the plasma carnitine level can be maintained by efficient renal reabsorption.
Prognosis
In developed countries, the majority of the PCD cases are identified by newborn screening tests. Early diagnosis and prompt supplementation with L- carnitine has improved the prognosis.[3] The majority of PCD cases have a better outcome as long as L-carnitine therapy is maintained. However, this condition can be fatal if left untreated.[13] Sudden death could be the manifestation of undiagnosed patients.[6]
Complications
Generally, PCD is well tolerated with early diagnosis followed by prompt and aggressive management with L-carnitine. The clinical manifestations and severity of symptoms can be variable between patients with PCD. Undiagnosed or poorly controlled PCD may lead to severe complications. Hypoglycemic episodes can result in convulsions and brain damage.[15] Untreated episodes of hypoglycemia may progress to coma and death.[8] Metabolic abnormalities can cause hepatic dysfunction that also may progress to hepatic encephalopathy culminating in death.[15] Cardiac abnormalities include cardiomyopathy, which can be fatal, especially during the neonatal period or early childhood; left ventricular hypertrophy that can progress to dilated cardiomyopathy and subsequent decrease in left ventricular ejection fraction; pericardial effusion; and arrhythmias including the long QT syndrome.[15] Untreated asymptomatic adults are also at risk of the development of cardiac arrhythmias and sudden death.[3]
Deterrence and Patient Education
- Carnitine supplementation and prevention of hypoglycemia are the mainstays of PCD treatment. Catabolic states such as fasting should be avoided. Other inevitable catabolic states, such as intercurrent infections, should be recognized early. Additionally, metabolic decompensation should be aggressively managed with intravenous dextrose. Patients with PCD should continue L-carnitine therapy. Hypoglycemic episodes and sudden cardiac death have been reported in patients with poor compliance with L-carnitine therapy.[18]
- Patient and caregiver education on proper treatment, especially medication compliance is essential for the management of PCD. Untreated patients can have profound effects that may progress to death. Adult females who are planning to get pregnant should be informed that pregnancy is a metabolically challenging situation that needs to be evaluated and managed with both a metabolic and genetic specialist before conception.
- In patients with SCD, it is crucial to prevent hypoglycemic episodes by frequent feeding and avoid fasting during day time and continuous corn starch supply during the night.[3][17][3] Patients should consume high carbohydrates and low-fat diets that provide energy in the form of medium-chain triglycerides, which can enter into the mitochondria in the absence of a carnitine shuttle.[21]
Enhancing Healthcare Team Outcomes
- In the U.S., newborn screening identifies most cases of PCD. The disorder is best managed by an interprofessional team approach that involves a metabolic specialist and genetic counselor, pharmacist, dietician, pediatrician, neurologist, hepatologist, endocrinologist, and cardiologist. All patients must be treated right after being diagnosed, or else this disorder may be potentially fatal.
- In PCD, the patient and caregiver should be educated on the importance of medication compliance.
- Genetic counseling should be provided in patients with inborn errors of carnitine metabolism. Antenatal diagnosis, using chorionic villus sampling or amniocentesis, should be performed for high-risk pregnancies as the recurrent risk in each pregnancy is 25% due to their autosomal recessive inheritance.[14]
- Chronic management of FAO defects and carnitine shuttle disorders consists of avoiding fasting, frequent feedings, high carbohydrate, low fat, and carnitine supplementation. Potentially hepatotoxic medications such as valproate should be avoided.[21]
- Patients with carnitine deficiency disorders with the risk of hypoglycemia can be recommended to wear medical alert devices such as bracelets. This will help paramedics and emergency department physicians to quickly recognize and manage emergencies such as hypoglycemia in carnitine deficit states.