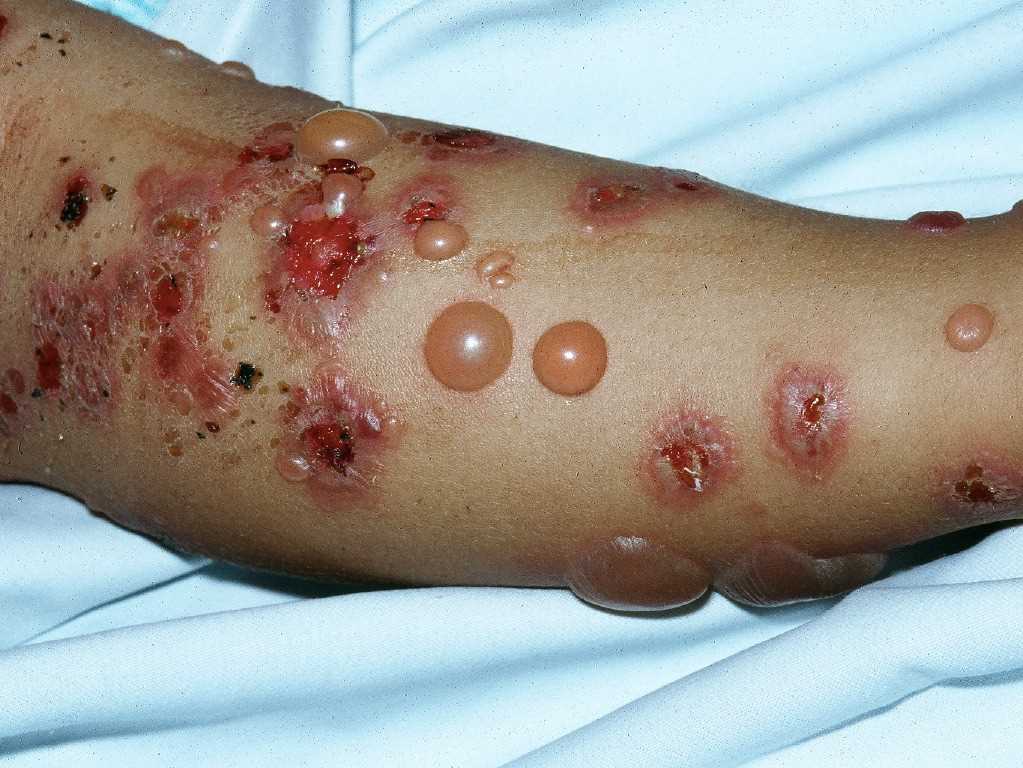
[1]
Vaillant L, Bernard P, Joly P, Prost C, Labeille B, Bedane C, Arbeille B, Thomine E, Bertrand P, Lok C, Roujeau JC. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. French Bullous Study Group. Archives of dermatology. 1998 Sep:134(9):1075-80
[PubMed PMID: 9762017]
[2]
Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, Vaillant L, D'Incan M, Plantin P, Bedane C, Young P, Bernard P, Bullous Diseases French Study Group. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. The New England journal of medicine. 2002 Jan 31:346(5):321-7
[PubMed PMID: 11821508]
[3]
Stavropoulos PG, Soura E, Antoniou C. Drug-induced pemphigoid: a review of the literature. Journal of the European Academy of Dermatology and Venereology : JEADV. 2014 Sep:28(9):1133-40. doi: 10.1111/jdv.12366. Epub 2014 Jan 10
[PubMed PMID: 24404939]
[6]
Giudice GJ, Emery DJ, Diaz LA. Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. The Journal of investigative dermatology. 1992 Sep:99(3):243-50
[PubMed PMID: 1324962]
[7]
Stanley JR, Tanaka T, Mueller S, Klaus-Kovtun V, Roop D. Isolation of complementary DNA for bullous pemphigoid antigen by use of patients' autoantibodies. The Journal of clinical investigation. 1988 Dec:82(6):1864-70
[PubMed PMID: 2461961]
[8]
Nishioka K,Hashimoto K,Katayama I,Sarashi C,Kubo T,Sano S, Eosinophilic spongiosis in bullous pemphigoid. Archives of dermatology. 1984 Sep
[PubMed PMID: 6383222]
[9]
Weigand DA, Clements MK. Direct immunofluorescence in bullous pemphigoid: effects of extent and location of lesions. Journal of the American Academy of Dermatology. 1989 Mar:20(3):437-40
[PubMed PMID: 2645321]
[10]
Mutasim DF, Adams BB. Immunofluorescence in dermatology. Journal of the American Academy of Dermatology. 2001 Dec:45(6):803-22; quiz 822-4
[PubMed PMID: 11712024]
[11]
Lazarova Z, Yancey KB. Reactivity of autoantibodies from patients with defined subepidermal bullous diseases against 1 mol/L salt-split skin. Specificity, sensitivity, and practical considerations. Journal of the American Academy of Dermatology. 1996 Sep:35(3 Pt 1):398-403
[PubMed PMID: 8784276]
[12]
Barnadas MA, Gelpi C, Curell R, de Moragas JM, Alomar A. Repeat direct immunofluorescence (DIF) test, using, 1 M NaCl treated skin, in the subepidermal autoimmune bullous diseases that contain IgG at the dermal epidermal junction. Journal of cutaneous pathology. 1999 Jan:26(1):37-41
[PubMed PMID: 10189243]
[13]
Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, Calobrisi SD. Bullous pemphigoid presenting as generalized pruritus: observations in six patients. International journal of dermatology. 1998 Jul:37(7):508-14
[PubMed PMID: 9679691]
[14]
Di Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clinics in dermatology. 2012 Jan-Feb:30(1):3-16. doi: 10.1016/j.clindermatol.2011.03.005. Epub
[PubMed PMID: 22137222]
[15]
Waisbourd-Zinman O, Ben-Amitai D, Cohen AD, Feinmesser M, Mimouni D, Adir-Shani A, Zlotkin M, Zvulunov A. Bullous pemphigoid in infancy: Clinical and epidemiologic characteristics. Journal of the American Academy of Dermatology. 2008 Jan:58(1):41-8
[PubMed PMID: 17945382]
[16]
Petronius D, Bergman R. Bullous pemphigoid in two young infants. Pediatric dermatology. 2002 Mar-Apr:19(2):119-21
[PubMed PMID: 11994172]
[17]
Fisler RE, Saeb M, Liang MG, Howard RM, McKee PH. Childhood bullous pemphigoid: a clinicopathologic study and review of the literature. The American Journal of dermatopathology. 2003 Jun:25(3):183-9
[PubMed PMID: 12775979]
[18]
Martinez-De Pablo MI, González-Enseñat MA, Vicente A, Gilaberte M, Mascaró JM Jr. Childhood bullous pemphigoid: clinical and immunological findings in a series of 4 cases. Archives of dermatology. 2007 Feb:143(2):215-20
[PubMed PMID: 17310001]
Level 3 (low-level) evidence
[19]
Chiavérini C, Hamel-Teillac D, Gilbert D, Prost Y. Absence of anti-BP180 antibodies in mothers of infants with bullous pemphigoid. The British journal of dermatology. 2006 May:154(5):839-43
[PubMed PMID: 16634883]
[20]
Ong E, Goldacre R, Hoang U, Sinclair R, Goldacre M. Associations between bullous pemphigoid and primary malignant cancers: an English national record linkage study, 1999-2011. Archives of dermatological research. 2014 Jan:306(1):75-80. doi: 10.1007/s00403-013-1399-5. Epub 2013 Aug 4
[PubMed PMID: 23912480]
[21]
Sakuma-Oyama Y, Powell AM, Oyama N, Albert S, Bhogal BS, Black MM. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. The British journal of dermatology. 2004 Jul:151(1):126-31
[PubMed PMID: 15270881]
[22]
Mariotti F, Grosso F, Terracina M, Ruffelli M, Cordiali-Fei P, Sera F, Zambruno G, Mastrogiacomo A, Di Zenzo G. Development of a novel ELISA system for detection of anti-BP180 IgG and characterization of autoantibody profile in bullous pemphigoid patients. The British journal of dermatology. 2004 Nov:151(5):1004-10
[PubMed PMID: 15541078]
[23]
Roujeau JC, Lok C, Bastuji-Garin S, Mhalla S, Enginger V, Bernard P. High risk of death in elderly patients with extensive bullous pemphigoid. Archives of dermatology. 1998 Apr:134(4):465-9
[PubMed PMID: 9554299]
[24]
Williams HC, Wojnarowska F, Kirtschig G, Mason J, Godec TR, Schmidt E, Chalmers JR, Childs M, Walton S, Harman K, Chapman A, Whitham D, Nunn AJ, UK Dermatology Clinical Trials Network BLISTER Study Group. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial. Lancet (London, England). 2017 Apr 22:389(10079):1630-1638. doi: 10.1016/S0140-6736(17)30560-3. Epub 2017 Mar 6
[PubMed PMID: 28279484]
Level 1 (high-level) evidence
[25]
Bilgiç Temel A, Bassorgun CI, Akman-Karakaş A, Alpsoy E, Uzun S. Successful Treatment of a Bullous Pemphigoid Patient with Rituximab Who Was Refractory to Corticosteroid and Omalizumab Treatments. Case reports in dermatology. 2017 Jan-Apr:9(1):38-44. doi: 10.1159/000452828. Epub 2017 Feb 10
[PubMed PMID: 28413387]
Level 3 (low-level) evidence
[26]
Fichel F, Barbe C, Joly P, Bedane C, Vabres P, Truchetet F, Aubin F, Michel C, Jegou J, Grange F, Antonicelli F, Bernard P. Clinical and immunologic factors associated with bullous pemphigoid relapse during the first year of treatment: a multicenter, prospective study. JAMA dermatology. 2014 Jan:150(1):25-33. doi: 10.1001/jamadermatol.2013.5757. Epub
[PubMed PMID: 24226428]
[27]
Pezzolo E, Naldi L. Epidemiology of major chronic inflammatory immune-related skin diseases in 2019. Expert review of clinical immunology. 2020 Feb:16(2):155-166. doi: 10.1080/1744666X.2020.1719833. Epub 2020 Jan 28
[PubMed PMID: 31962053]