Continuing Education Activity
Thin basement membrane disease or thin basement membrane nephropathy (TBMN) is a genetic disorder affecting the glomerular basement membrane. TBMN is characterized by microscopic hematuria, usually with normal kidney function and without proteinuria. TBMN is caused by mutations in genes encoding type IV collagen, resulting in the extensive thinning of the glomerular basement membrane. While TBMN does have overlapping features with Alport syndrome, the course and prognosis of the two diseases differ significantly, and each requires different management strategies. A definitive diagnosis of TMBN is needed to optimize patient outcomes. This activity reviews the pathogenesis, evaluation, and management of TBMN and discusses the interprofessional team's role in evaluating and treating patients with this condition.
Objectives:
- Correlate the pathogenesis of thin basement membrane nephropathy with the typical clinical signs and symptoms of the disease.
- Employ best practices when evaluating the severity of thin basement membrane nephropathy in affected patients.
- Assess and manage the complications of thin basement membrane nephropathy.
- Collaborate with interprofessional team members to improve outcomes for patients with thin basement membrane nephropathy.
Introduction
Thin basement membrane nephropathy (TBMN), also known as thin basement membrane disease, is one of the most common but under-recognized causes of glomerular bleeding in both children and adults.[1] It is a genetic and often familial disorder caused by mutations in the genes encoding various chains of type IV collagen, which is the main component of the glomerular basement membrane (GBM).[2]
TBMN was previously known as benign familial hematuria, but this term is no longer favored due to improved insights into the genetic etiologies and variations in the clinical presentation of the condition.[3][4]
The condition is characterized by diffuse thinning of the GBM on renal biopsy and clinically presents as isolated microscopic hematuria in most cases.[5][6] However, other findings have also been identified, including proteinuria, hypertension, and varying degrees of kidney function impairment.[3][7][8]
TBMN has features that overlap with Alport syndrome, as genetic mutations of type IV collagen genes cause both conditions.[3][9] However, unlike Alport syndrome, particularly its most common X-linked variant, TBMN has no extra-renal features.[10]
Etiology
Mutations in COL4A3 and COL4A4 cause thin basement membrane nephropathy. These genes are located on chromosome 2 at 2q35-37 and encode for 2 different chains of type IV collagen.[2][3][11] The mutations are heterozygous, or single allele mutations, and can have many variations, including splice variants or missense or frameshift mutations.[2]
One-half to two-thirds of patients with TBMN have an identifiable family history, with inheritance in an autosomal dominant pattern.[3][9] Patients without an identifiable family history may have a de novo mutation. This disease also demonstrated incomplete penetrance, resulting in a lack of supportive family history.[1][11]
Epidemiology
Thin basement membrane nephropathy is a common cause of glomerular hematuria in children and adults, and the age at presentation will vary. The median age at presentation for patients diagnosed in adulthood is 37 years.[1] Most case series demonstrate that TBMN is more common in females without predilection for any race or ethnicity.[1][12]
The exact prevalence of the condition is difficult to estimate due to the variable clinical presentation of TBMN. Many cases are thought to be unrecognized. However, estimates from a biopsy series indicate the incidence of thin basement membrane morphology is present in about 5% to 9% of the general population.[13] More conservative estimates place the incidence of TBMN between 1% and 2%.[1][11][14]
Pathophysiology
The GBM is the acellular middle layer of the glomerular filtration barrier between 2 cellular layers of endothelial cells on one side and epithelial foot processes on the other.[15][16] The GBM is composed of extracellular matrix protein, the most abundant of which is type IV collagen.[15][16]
Type IV collagen in the GBM of adults is formed by the complex assembly of three chains, the α-3, α-4, and α-5 chains, to form a triple helix.[2][17] A distinct gene encodes each α-chain protein. The α-5 chain is encoded by COL4A5 on the X-chromosome. The α-3 and α-4 chains are encoded by COL4A3 and COL4A4 on chromosome 2, respectively.[2][17] The type IV collagen chains α-1 and α-2 are seen typically only during embryonic development.[2][17]
Heterozygous mutations in COL4A3 and COL4A4 are the most commonly reported genetic abnormalities in TBMN.[1][3][9] These mutations result in abnormal folding and assembly of the collagen chain resulting in early degradation. This degradation prevents the normal developmental switch from the α-1 and α-2 chains to the α-3, α-4, and α-5 chains.[2] The embryonic forms of type IV collagen chains are more susceptible to degradation, resulting in structural changes and thinning of the GBM.[2]
The thin GBM is fragile and sensitive to disruption by proteases, causing transient, localized ruptures of the GBM. These protease-induced ruptures of the glomerular filtration barrier allow the egress of red blood cells (RBCs) from inside the capillary space into the Bowman space of the glomerulus. Once the RBCs enter the Bowman space, they appear in the urine as hematuria.[18][19]
A similar pathogenic process is seen in Alport syndrome, most commonly due to mutations in COL4A5 on the X-chromosome.[3][10] However, Alport syndrome has also been described in patients with mutations in COL4A3 and COL4A4, inherited in autosomal dominant or autosomal recessive patterns.[3][10][11][20] Therefore, the distinction between TBMN and Alport syndrome may be difficult, and considerable overlap in clinical presentation can exist. Alport syndrome and TBMN are now considered on a spectrum of type IV collagen disorders.[3][21]
Histopathology
The pathognomonic finding of TBMN is uniform thinning of the GBM revealed by electron microscopy of a kidney biopsy.[4][5][6]
Light Microscopy
No specific findings are typically observed by light microscopy in cases of TBMN. The glomeruli may appear normal or demonstrate mild mesangial cell proliferation or mild mesangial matrix expansion. Rarely, slight attenuation of the GBM indicative of thinning can be seen with Jones methenamine silver or periodic acid-Schiff staining (see Image. Light Microscopy of Renal Parenchyma in Thin Basement Membrane Nephropathy).[4]
Global or focal segmental glomerulosclerosis is not typically associated with TBMN but, if present, may indicate additional pathologies or nonspecific manifestations of the progression of kidney disease. Similarlas interstitial fibrosis and tubular atrophy are nonspecific indicators of progressive kidney dysfunction.[1][4][5]
Immunofluorescence
Immunofluorescence is usually negative. Nonspecific staining of mesangium for IgM and C3 can sometimes be seen, and even more rarely, staining for IgG or IgA will be positive.[1][4][6]
Unlike X-linked Alport syndrome, immunohistochemistry for collagen staining does not always help in the diagnosis of TBMN. While X-linked Alport syndrome biopsies consistently reveal a lack of staining for the α-5 chain, the autosomal forms of the type IV collagen disorders involving α-3 and α-4 chains can demonstrate a normal staining pattern depending on the type of mutations.[5]
Electron Microscopy
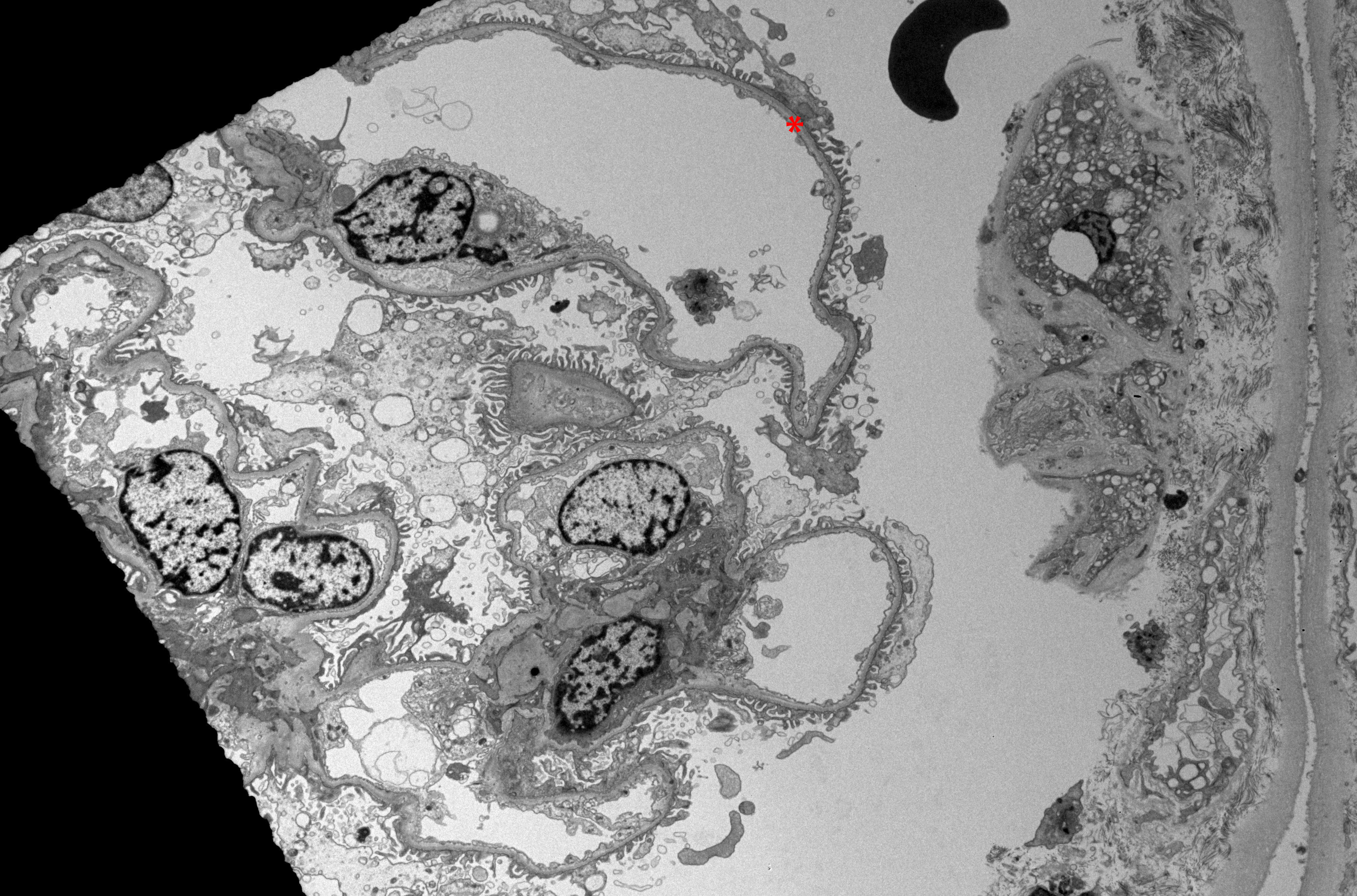
The pathognomonic feature of TBMN, uniform and extensive (>50%) thinning of the GBM, is visualized with electron microscopy (see Image. Electron Microscopy of Renal Parenchyma in Thin Basement Membrane Nephropathy).[1] [22] Importantly, electron microscopy features of progressive irregularity in the thickness of the GBM with splitting and layering that are seen in Alport syndrome are absent in TBMN.[5][10][23][24]
The thickness of the GBM normally varies with age. The GBM is approximately 150 nm thick at birth and increases throughout childhood. By age 11 years, GBM thickness has approached adult measurements. The mean normal GBM thickness in adult males is 370 ± 50 nm, and in adult females is 320 ± 50 nm.[4][22] The World Health Organization proposed the threshold most commonly used to determine a thin basement membrane which is 250 nm for adults and 180 nm for children between the ages of 2 and 11.[4][22]
History and Physical
Most cases of TBMN initially present with isolated asymptomatic microscopic hematuria identified incidentally. The medical history and physical examination in these patients are typically otherwise unremarkable. A familial history of hematuria may or may not be present.
Gross hematuria, or "cola-colored" urine, frequently seen in other glomerulonephropathies, is less common in TBMN; gross hematuria is seen in just 5% to 30% of cases. If present, gross hematuria is more common in children with TBMN.[1][4] Proteinuria is uncommon regardless of age and is usually seen in <10% of cases. When present, proteinuria is sub-nephrotic, defined as <3000 mg protein/24 hours.[1][4] There are usually no extra-renal manifestations in patients with TBMN.[1][4][22]
The medical history and physical examination should focus on identifying a potential or obvious cause for the microscopic hematuria. The medical history must focus on ruling out more common causes of microscopic hematuria, including urological etiologies and nephrolithiasis. A thorough review of systems can help rule out systemic manifestations of autoimmune diseases, such as systemic lupus erythematosus, that can present with microscopic hematuria.
Most patients with TBMN will report a family history of hematuria without the development of end-stage kidney disease (ESKD). Such a history with a negative urological workup would strongly suggest the diagnosis of TBMN. A specific history of hearing loss and ocular lesions can help differentiate TBMN from Alport syndrome.[3][10]
The family medical history is crucial to evaluating TBMN to establish potential familial cases, differentiate TBMN from X-linked Alport syndrome, and establish a prognosis.[3] Questioning must include inquiry into other instances of microscopic or gross hematuria, hearing or vision loss, and, most importantly, progressive kidney disease or dialysis dependence in other family members.
The physical examination must focus on blood pressure measurement, determining the presence of edema suggestive of proteinuria, careful assessment of vision and hearing, and determining the presence of other systemic signs such as rash or joint involvement that can point towards other potential causes of glomerular hematuria.
Evaluation
Initial Diagnostic Evaluation
The initial diagnostic evaluation of suspected TBMN includes urinalysis, microscopic examination of urinary sediment, and laboratory evaluation of kidney function.
In patients with TBMN, a dipstick urinalysis usually reveals microscopic hematuria without proteinuria.[1][4][22] The urine protein must be quantified if proteinuria is noted on dipstick testing. Urine protein quantification is most commonly performed using a spot urine sample and measuring the urine protein-to-creatinine ratio. A protein/creatinine ratio of <0.2 is considered normal, and 0.3 or higher is abnormal. A 24-hour urine protein quantification is considered the gold standard for determining proteinuria but is cumbersome.[25][26]
Microscopic examination of the urine sediment allows for identifying dysmorphic RBCs or RBC casts that are pathognomonic of glomerular hematuria.[27] The RBC dysmorphia is presumptively due to passage through the gaps in the GBM, and RBC casts are formed when RBCs enter the tubular lumen and are subsequently trapped in the tubular protein matrix.[28] However, the absence of dysmorphic RBCs or RBC casts does not rule out glomerular hematuria.
Kidney function is evaluated with serum creatinine and blood urea nitrogen. Measurement of serum albumin should be performed to rule out nephrotic syndrome if proteinuria is present.
Renal Biopsy
Beyond the initial investigations, the primary decision that will need to be made when evaluating TBMN is whether a kidney biopsy should be performed. When the presentation and course of the disease are benign, characterized by isolated microscopic hematuria without proteinuria, hypertension, or laboratory evidence of kidney dysfunction, and there is no family history of progressive kidney dysfunction or ESKD, a biopsy is not recommended, and the diagnosis is inferred clinically.[1][4]
However, a kidney biopsy can be considered in the following circumstances:
- Presence of other features, such as laboratory evidence of kidney injury, proteinuria, or hypertension, particularly in individuals younger than 50 years.
- Presence of extra-renal manifestations such as hearing and ocular defects or family history suggestive of kidney disease such as Alport syndrome, particularly X-linked Alport syndrome.
- If the individual is considering a kidney donation.
Genetic Testing
Genetic testing is usually not required to diagnose TBMN.[9] The genes involved in the pathogenesis of TBMN are COL4A3 and COL4A4 on chromosome 2.[1][2] The rate of detection of mutations is low in TBMN in patients with the sole clinical manifestation of microscopic hematuria.[1] The likelihood of detecting a pathogenic variant is higher if there is kidney function impairment or an associated family history.[29]
In patients with biopsy-proven thin GBM but associated kidney dysfunction and a family history suggestive of X-linked inheritance, screening for COL4A5 mutations to exclude X-linked Alport syndrome is more important.[9][29]
Treatment / Management
No specific, evidence-based management strategies for TBMN are available.[1][9] Most patients with TBMN with isolated microscopic hematuria have a benign clinical course and do not require any specific therapy.[4]
Once the diagnosis is made, management involves clinical monitoring for the development of hypertension with regular blood pressure measurements and laboratory studies, including measurement of serum creatinine and blood urea nitrogen, and urinalyses with urine protein-to-creatinine ratio for the development of renal impairment and proteinuria. The primary care practitioner can carry out these routine studies with referrals as needed.
If proteinuria or hypertension is noted, management strategies include:
- Blood pressure management with a goal of less than 130/80 mmHg, consistent with the 2017 American College of Cardiology/American Heart Association Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults.[30]
- Minimizing salt intake, with a target sodium intake of less than 2000 mg daily.
- Moderate-intensity physical activity for a total duration of at least 150 minutes per week.
- Pharmacological therapy is recommended with maximum tolerated doses of renin-angiotensin-system inhibitors with angiotensin-converting enzyme inhibitor (ACE-inhibitor) or angiotensin receptor blocker (ARB).[1][9][22]
There is no role for immunosuppression in the management of TBMN.
Once the patient is started on treatment for hypertension or proteinuria, or if kidney dysfunction is noted, more frequent monitoring and management of associated complications, including progression of chronic kidney disease (CKD), anemia due to CKD, CKD-related mineral bone disorder, and ESKD, are needed. The frequency of monitoring varies based on blood pressure control, degree of proteinuria, and degree of kidney dysfunction or stage of CKD. This is best carried out under the purview of a nephrologist.
Genetic Counseling
Counseling for patients with TBMN and their families is recommended. Counseling should include education regarding the diagnosis, the likely inherited nature of the disease, and the need for monitoring for the presence of microscopic hematuria, along with proteinuria, hypertension, and kidney function impairment.[9][29]
Renal Transplant Donation
Historically, it was suggested that potential renal transplant donors with TBMN might be acceptable as some organ recipients have maintained kidney function without significant problems or complications.[31] However, as the overlap between TBMN and autosomal forms of Alport syndrome is being delineated and the risk of progressive kidney disease is higher than previously thought, it is now recommended that individuals with a heterozygous mutation of COL4A3 or COL4A4 not be kidney donors.[29]
Differential Diagnosis
Alport Syndrome
The condition most closely related to TBMN that requires differentiation is Alport syndrome. It is important to attempt to differentiate between these two conditions due to differences in prognosis.[3][10][21][23][24] The salient differences between the two conditions include:
- Mutations and Inheritance: The most well-recognized form of Alport syndrome is inherited in an X-linked fashion and is caused by mutations in COL4A5. However, autosomal recessive and autosomal dominant patterns of inheritance are also seen in Alport syndrome due to homozygous or heterozygous mutations in COL4A3 and COL4A4. TBMN is most commonly inherited in an autosomal dominant pattern due to heterozygous mutations in COL4A3 or COL4A4.
- Histopathology: The isolated finding of TBMN revealed by renal biopsy is electron microscopic evidence of uniform and >50% thinning of the GBM.[18][32][33] The pathognomonic biopsy feature of Alport syndrome is progressive irregularity and uneven thickness of the GBM with electron microscopic evidence of splitting and layering in the presence of small, dense, "breadcrumb" inclusions between the layers.[34][35][36] A combination of electron microscopy and immunohistology for α-3 and α-5 collagen subtypes will permit a definitive diagnosis from a renal biopsy, even in difficult cases.[24]
- Presentation: X-linked Alport syndrome is commonly seen in males and presents at a very young age. The typical presentation is characterized by hearing loss, vision changes, and a high risk of progression to ESKD. Female carriers of X-linked Alport syndrome have a milder clinical presentation and lower risk of progression to ESKD. The autosomal dominant forms of Alport syndrome and TBMN present similarly, do not usually have extra-renal features, and the risk of progression to ESKD depends on the presence of proteinuria and a positive family history of renal disease.
- Management: Neither Alport syndrome nor TBMN are curable. The only approved pharmacological therapy for either condition is ACE inhibitors or ARBs in the setting of proteinuria. There is no role for immunosuppression in either condition. Novel agents currently being evaluated but not yet approved for the treatment of Alport syndrome include bardoxolone methyl, a semi-synthetic triterpenoid molecule that activates the transcription factor nuclear factor erythroid 2-related factor 2 (CARDINAL trial), sparsentan, a dual endothelin type A receptor and angiotensin II type 1 receptor antagonist (EPPIK trial), and atrasentan, an endothelin type-A receptor antagonist (AFFINITY trial).[37] No agents are currently being studied for the treatment of TBMN.
- Prognosis: The X-linked form of Alport syndrome has a poor prognosis, with about 50% of affected males reaching ESKD by age 30 and about 90% by age 40. Females with the X-linked form of Alport syndrome have a better prognosis, with a 25% lifetime risk of progression. The autosomal recessive forms of Alport syndrome also have a poor prognosis, with most patients reaching ESKD by age 30. The autosomal dominant forms of Alport syndrome and TBMN nephropathy have similar prognoses with a <1% estimated risk of progression to ESKD in the absence of factors such as proteinuria and significant family history of progression. In the presence of these factors, the risk for progression to ESKD increases up to 20%.
In summary, due to their shared molecular pathophysiological basis and overlapping clinical presentation, it has been proposed that both Alport syndrome and TBMN be classified as forms of type IV collagen disorders.[3]
Other Conditions
Other conditions that form the differential diagnoses for TBMN include:
- IgA nephropathy [38]
- Postinfectious glomerulonephritis [39][40]
- Lupus nephritis [41]
- Medullary sponge kidney [42]
Urological conditions such as malignancies and nephrolithiasis should always be considered in the differential diagnoses of isolated microscopic hematuria. Patients less than 50 years of age with asymptomatic microscopic hematuria without an identified urological cause should consider nephrology consultation and evaluation. This is particularly important if the patient demonstrates persistent proteinuria, abnormal serum creatinine, red cell casts or dysmorphic RBCs on microscopic urinalysis, worsening hypertension or edema, or a family history of kidney disease.
Prognosis
The long-term prognosis in most patients with TBMN is very good.[1][22] The condition usually does not lead to the progression of kidney disease or ESKD.
However, as the genetic basis of TBMN and its overlap with other type IV collagen disorders that demonstrate mutations in COL4A3 and COL4A4, such as Alport syndrome, variants of IgA nephropathy, and focal segmental glomerulosclerosis have been elucidated in more detail, TBMN is no longer considered a universally benign condition that requires no monitoring.[3][21] Therefore, the term "benign familial hematuria" is no longer recommended for this condition, and "thin basement membrane nephropathy" or "thin basement membrane disease" are preferred.[3]
The estimated risk of progression to ESKD in those with heterozygous mutations of COL4A3 or COL4A4 inherited in an autosomal dominant fashion, such as in TBMN, is <1% in the absence of associated risk factors.[3] However, in those with risk factors such as the evidence of proteinuria, the presence or progression of renal dysfunction on laboratory studies, certain biopsy features including FSGS, thickening and lamellation of the GBM, and a family history of progression of kidney disease, the estimated risk of progression to ESKD is up to 20%.[3]
Complications
The complications of TBMN include hypertension, proteinuria, and ESKD. These complications are rare. Of equal importance to recognize are complications related to anxiety due to the diagnosis, especially in young individuals, the uncertainty and ambiguity of genetic testing without proper indication or counseling, and the potential complications related to investigations such as kidney biopsy.
Obtaining a comprehensive medical history that includes a family history, genetic counseling, and careful selection of patients for genetic testing and biopsy can help mitigate these complications.
Deterrence and Patient Education
TBMN is a genetic condition, and therefore there are no preventive measures as part of its management. Patient education must include information regarding the genetic basis of the condition and its autosomal dominant inheritance, although rare de novo cases can be seen. Education must also focus on the need for monitoring for the development of hypertension, proteinuria, and worsening kidney function. Genetic counseling is an essential component of patient education for TBMN. While reassuring patients regarding the predominantly benign course of the disease is important to prevent unnecessary anxiety, it is equally important to identify and educate patients regarding the risk factors for progression to end-stage renal disease and dialysis.
Pearls and Other Issues
Patients with unexplained persistent microscopic hematuria after a urological evaluation should be referred to a nephrologist if they also have any other signs of possible renal disease, such as abnormal serum creatinine, red cell casts, dysmorphic RBCs on microscopic urinalysis, proteinuria, progressive hypertension, edema, or a family history of kidney disease.
Failing to find dysmorphic RBCs or RBC casts on urinalysis does not rule out a glomerular source for hematuria, even if the urological workup, including computed tomography (CT) urogram and cystoscopy, is completely negative. Although usually benign and not requiring treatment, if TBMN has associated features such as proteinuria or hypertension, management with angiotensin-converting enzyme inhibitors or angiotensin-receptor-blockers is recommended. It is important to differentiate TBMN, which is generally benign, from Alport syndrome, where renal function progressively declines. This differentiation may require a renal biopsy and electron microscopy.[23][24]
Enhancing Healthcare Team Outcomes
TBMN is a genetic condition that affects the kidneys alone without extra-kidney manifestations. As such, a nephrologist, both adult and pediatric, is vital in managing TBMN, which can present in children and young adults.
Often, patients with microscopic hematuria are initially evaluated by urologists to rule out the more common urological causes of hematuria. Coordination between the urologist and nephrologist is essential for the continued evaluation of patients, particularly those younger than 50, with asymptomatic microscopic hematuria and negative urological workups.
However, well-rounded management of patients with TBMN requires a multidisciplinary team of interprofessional healthcare practitioners. This team includes primary care practitioners for the initial evaluation of microscopic hematuria and continued monitoring of patients for the development of risk factors such as proteinuria, hypertension, or kidney dysfunction, geneticists for appropriate counseling and testing of both the patients and their family members, and pathologists to identify histopathology appropriately.
If progression to ESKD is expected, patients require further education and preparation for kidney replacement therapy. At such times, additional interprofessional team members, including nurses, dieticians, and social workers, play an essential role in managing these patients.
Finally, when individuals considering kidney donation are diagnosed with TBMN, the transplant nephrologist, and the multidisciplinary living donation team must counsel the individuals regarding their risk of kidney donation and the need for monitoring in the future.
Therefore, collaboration among the interprofessional team of healthcare providers is essential for the appropriate management of TBMN, which is one of the common causes of glomerular hematuria that can be mischaracterized as entirely benign but needs monitoring to identify those at risk for progressing to ESKD.[3][21]