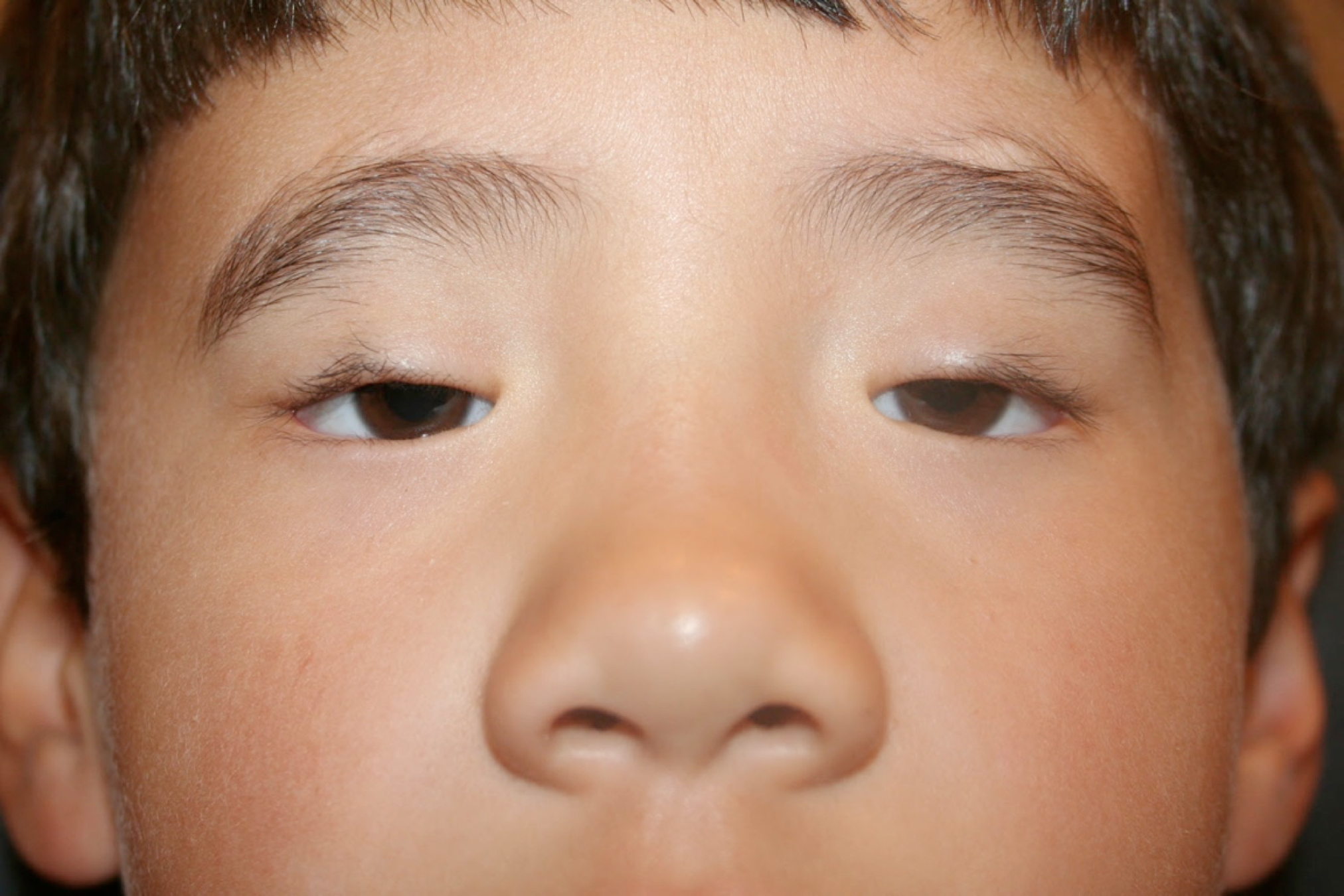
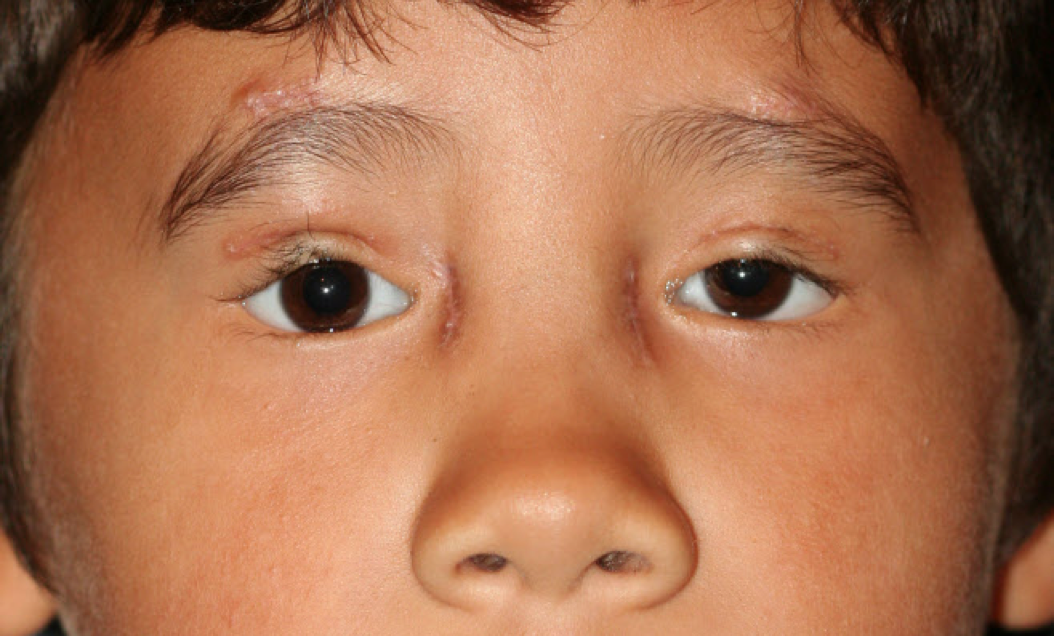
[1]
Méjécase C, Nigam C, Moosajee M, Bladen JC. The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome. Genes. 2021 Mar 4:12(3):. doi: 10.3390/genes12030364. Epub 2021 Mar 4
[PubMed PMID: 33806295]
Level 3 (low-level) evidence
[2]
Allen CE, Rubin PA. Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES): clinical manifestation and treatment. International ophthalmology clinics. 2008 Spring:48(2):15-23. doi: 10.1097/IIO.0b013e3181694eee. Epub
[PubMed PMID: 18427257]
[3]
Gupta AK, Gupta DC, Khan SA, Razi SM. Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES) Type 1 in an Indian Family. Journal of the ASEAN Federation of Endocrine Societies. 2017:32(1):68-71. doi: 10.15605/jafes.032.01.13. Epub 2017 May 9
[PubMed PMID: 33442089]
[4]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Verdin H, Matton C, De Baere E. Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome. GeneReviews(®). 1993:():
[PubMed PMID: 20301614]
[5]
Yu Y, Ji M, Xu W, Zhang L, Qi M, Shu J. Confrontment and solution to gonadotropin resistance and low oocyte retrieval in in vitro fertilization for type I BPES: a case series with review of literature. Journal of ovarian research. 2021 Oct 28:14(1):143. doi: 10.1186/s13048-021-00900-2. Epub 2021 Oct 28
[PubMed PMID: 34711234]
Level 2 (mid-level) evidence
[6]
Wang S, Ge S, Zhuang A. A Novel Forkhead Box L2 Missense Mutation, c.1068G}C, in a Chinese Family With Blepharophimosis/Ptosis/ Epicanthus Inversus Syndrome. The Journal of craniofacial surgery. 2022 May 1:33(3):e238-e240. doi: 10.1097/SCS.0000000000008042. Epub 2021 Aug 9
[PubMed PMID: 34374675]
[7]
Crisponi L, Deiana M, Loi A, Chiappe F, Uda M, Amati P, Bisceglia L, Zelante L, Nagaraja R, Porcu S, Ristaldi MS, Marzella R, Rocchi M, Nicolino M, Lienhardt-Roussie A, Nivelon A, Verloes A, Schlessinger D, Gasparini P, Bonneau D, Cao A, Pilia G. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nature genetics. 2001 Feb:27(2):159-66
[PubMed PMID: 11175783]
[8]
Beysen D, De Paepe A, De Baere E. FOXL2 mutations and genomic rearrangements in BPES. Human mutation. 2009 Feb:30(2):158-69. doi: 10.1002/humu.20807. Epub
[PubMed PMID: 18726931]
[9]
Tucker EJ. The Genetics and Biology of FOXL2. Sexual development : genetics, molecular biology, evolution, endocrinology, embryology, and pathology of sex determination and differentiation. 2022:16(2-3):184-193. doi: 10.1159/000519836. Epub 2021 Nov 2
[PubMed PMID: 34727551]
[10]
De Baere E, Beysen D, Oley C, Lorenz B, Cocquet J, De Sutter P, Devriendt K, Dixon M, Fellous M, Fryns JP, Garza A, Jonsrud C, Koivisto PA, Krause A, Leroy BP, Meire F, Plomp A, Van Maldergem L, De Paepe A, Veitia R, Messiaen L. FOXL2 and BPES: mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. American journal of human genetics. 2003 Feb:72(2):478-87
[PubMed PMID: 12529855]
[11]
Lin ZB, Chen ZJ, Yang H, Ding XR, Li J, Pan AP, Sun HS, Yu AY, Chen SH. Expanded phenotypic spectrum of FOXL2 Variant c.672_701dup revealed by whole-exome sequencing in a rare blepharophimosis, ptosis, and epicanthus inversus syndrome family. BMC ophthalmology. 2023 Nov 7:23(1):446. doi: 10.1186/s12886-023-03189-5. Epub 2023 Nov 7
[PubMed PMID: 37932670]
[12]
Shen Q, Zhao X, Ji Y, Chai P. Deletion of cis-regulatory Element in FOXL2 Promoter in a Chinese Family of Type II Blepharophimosis-ptosis-epicanthus Inversus Syndrome with Polydactyly. The Journal of craniofacial surgery. 2024 Jan-Feb 01:35(1):e52-e56. doi: 10.1097/SCS.0000000000009801. Epub 2023 Nov 8
[PubMed PMID: 37938073]
[13]
Fuller PJ, Nguyen T, Alexiadis M, Chu S. FOXL2(C134W) : much ado about something!(†). The Journal of pathology. 2022 Jan:256(1):1-3. doi: 10.1002/path.5816. Epub 2021 Nov 18
[PubMed PMID: 34687235]
[14]
França MM, Mendonca BB. Genetics of ovarian insufficiency and defects of folliculogenesis. Best practice & research. Clinical endocrinology & metabolism. 2022 Jan:36(1):101594. doi: 10.1016/j.beem.2021.101594. Epub 2021 Oct 14
[PubMed PMID: 34794894]
[15]
Chawla B, Bhadange Y, Dada R, Kumar M, Sharma S, Bajaj MS, Pushker N, Chandra M, Ghose S. Clinical, radiologic, and genetic features in blepharophimosis, ptosis, and epicanthus inversus syndrome in the Indian population. Investigative ophthalmology & visual science. 2013 Apr 26:54(4):2985-91. doi: 10.1167/iovs.13-11794. Epub 2013 Apr 26
[PubMed PMID: 23513057]
[16]
Krastinova D, Jasinski MA. Orbitoblepharophimosis syndrome: a 16-year perspective. Plastic and reconstructive surgery. 2003 Mar:111(3):987-99
[PubMed PMID: 12621168]
Level 3 (low-level) evidence
[17]
Beckingsale PS, Sullivan TJ, Wong VA, Oley C. Blepharophimosis: a recommendation for early surgery in patients with severe ptosis. Clinical & experimental ophthalmology. 2003 Apr:31(2):138-42
[PubMed PMID: 12648048]
[18]
Dawson EL, Hardy TG, Collin JR, Lee JP. The incidence of strabismus and refractive error in patients with blepharophimosis, ptosis and epicanthus inversus syndrome (BPES). Strabismus. 2003 Sep:11(3):173-7
[PubMed PMID: 14710475]
[19]
Liu CY. Wakayama Symposium: Notch-FoxL2-α-SMA axis in eyelid levator muscle development and congenital blepharophimosis. The ocular surface. 2012 Oct:10(4):221-3. doi: 10.1016/j.jtos.2012.07.003. Epub 2012 Jul 25
[PubMed PMID: 23084143]
[20]
Heude É, Bellessort B, Fontaine A, Hamazaki M, Treier AC, Treier M, Levi G, Narboux-Nême N. Etiology of craniofacial malformations in mouse models of blepharophimosis, ptosis and epicanthus inversus syndrome. Human molecular genetics. 2015 Mar 15:24(6):1670-81. doi: 10.1093/hmg/ddu579. Epub 2014 Nov 21
[PubMed PMID: 25416281]
[21]
Harris SE, Chand AL, Winship IM, Gersak K, Aittomäki K, Shelling AN. Identification of novel mutations in FOXL2 associated with premature ovarian failure. Molecular human reproduction. 2002 Aug:8(8):729-33
[PubMed PMID: 12149404]
[22]
Yang XW, He WB, Gong F, Li W, Li XR, Zhong CG, Lu GX, Lin G, Du J, Tan YQ. Novel FOXL2 mutations cause blepharophimosis-ptosis-epicanthus inversus syndrome with premature ovarian insufficiency. Molecular genetics & genomic medicine. 2018 Mar:6(2):261-267. doi: 10.1002/mgg3.366. Epub 2018 Jan 29
[PubMed PMID: 29378385]
[23]
Xue M, Zheng J, Zhou Q, Hejtmancik JF, Wang Y, Li S. Novel FOXL2 mutations in two Chinese families with blepharophimosis-ptosis-epicanthus inversus syndrome. BMC medical genetics. 2015 Sep 1:16():73. doi: 10.1186/s12881-015-0217-7. Epub 2015 Sep 1
[PubMed PMID: 26323275]
[24]
Wen F, Ding Y, Wang M, Du J, Zhang S, Kee K. FOXL2 and NR5A1 induce human fibroblasts into steroidogenic ovarian granulosa-like cells. Cell proliferation. 2024 May:57(5):e13589. doi: 10.1111/cpr.13589. Epub 2024 Jan 8
[PubMed PMID: 38192172]
[25]
JOHNSON CC. SURGICAL REPAIR OF THE SYNDROME OF EPICANTHUS INVERSUS, BLEPHAROPHIMOSIS AND PTOSIS. Archives of ophthalmology (Chicago, Ill. : 1960). 1964 Apr:71():510-6
[PubMed PMID: 14109036]
[26]
Beaconsfield M, Walker JW, Collin JR. Visual development in the blepharophimosis syndrome. The British journal of ophthalmology. 1991 Dec:75(12):746-8
[PubMed PMID: 1768667]
[27]
KLEIN M. Hereditary bilateral ptosis and blepharophimosis associated with other developmental abnormalities of the outer eye. Proceedings of the Royal Society of Medicine. 1950 Dec:43(12):1025-6
[PubMed PMID: 14808191]
[28]
Kohn R, Romano PE. Blepharoptosis, blepharophimosis, epicanthus inversus, and telecanthus--a syndrome with no name. American journal of ophthalmology. 1971 Sep:72(3):625-32
[PubMed PMID: 5568616]
[30]
JOHNSON CC. Operations for epicanthus and blepharophimosis; an evaluation and a method for shortening the medial canthal ligament. American journal of ophthalmology. 1956 Jan:41(1):71-9
[PubMed PMID: 13275546]
[31]
Lewis SR, Arons MS, Lynch JB, Blocker TG Jr. The congenital eyelid syndrome. Plastic and reconstructive surgery. 1967 Mar:39(3):271-7
[PubMed PMID: 5336149]
[33]
Garden JW. Blepharophimosis, ptosis, epicanthus inversus and lacrimal stenosis. American journal of ophthalmology. 1969 Jan:67(1):153-4
[PubMed PMID: 5782860]
[34]
Choi KH, Kyung S, Oh SY. The factors influencing visual development in blepharophimosis-ptosis-epicanthus inversus syndrome. Journal of pediatric ophthalmology and strabismus. 2006 Sep-Oct:43(5):285-8. doi: 10.3928/01913913-20060901-03. Epub
[PubMed PMID: 17022162]
[35]
Landau Prat D, Nguyen BJ, Strong A, Katowitz WR, Katowitz JA. "Blepharophimosis-plus" syndromes: Frequency of systemic genetic disorders that also include blepharophimosis. Clinical & experimental ophthalmology. 2021 Jul:49(5):448-453. doi: 10.1111/ceo.13933. Epub 2021 Apr 29
[PubMed PMID: 33882191]
[36]
Bertini V, Valetto A, Baldinotti F, Azzarà A, Cambi F, Toschi B, Giacomina A, Gatti GL, Gana S, Caligo MA, Bertelloni S. Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome: New Report with a 197-kb Deletion Upstream of FOXL2 and Review of the Literature. Molecular syndromology. 2019 May:10(3):147-153. doi: 10.1159/000497092. Epub 2019 Mar 20
[PubMed PMID: 31191203]
[37]
Zhuang J, Zeng S, Wang Y, Jiang Y. [Clinical and genetic analysis of a patient with 10q26.3 microdeletion in conjunct with 18q22.3q23 microduplication]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics. 2022 Dec 10:39(12):1415-1418. doi: 10.3760/cma.j.cn511374-20211126-00943. Epub
[PubMed PMID: 36453971]
[38]
Chen M, Jiang H, Zhang C. Selected Genetic Factors Associated with Primary Ovarian Insufficiency. International journal of molecular sciences. 2023 Feb 23:24(5):. doi: 10.3390/ijms24054423. Epub 2023 Feb 23
[PubMed PMID: 36901862]
[39]
Zeng L, Zhang Y, Lin L, Dong X, Li L. [Genetic testing and prenatal diagnosis for a family with 10q22.3q23.2 microdeletion]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics. 2021 Aug 10:38(8):768-770. doi: 10.3760/cma.j.cn511374-20200608-00419. Epub
[PubMed PMID: 34365621]
[40]
Yang Y, Yang C, Zhu Y, Chen H, Zhao R, He X, Tao L, Wang P, Zhou L, Zhao L, Tu M, Dong Z, Chen H, Xie Z. Intragenic and extragenic disruptions of FOXL2 mapped by whole genome low-coverage sequencing in two BPES families with chromosome reciprocal translocation. Genomics. 2014 Sep:104(3):170-6. doi: 10.1016/j.ygeno.2014.07.010. Epub 2014 Jul 30
[PubMed PMID: 25086333]
[41]
Huhtaniemi I, Hovatta O, La Marca A, Livera G, Monniaux D, Persani L, Heddar A, Jarzabek K, Laisk-Podar T, Salumets A, Tapanainen JS, Veitia RA, Visser JA, Wieacker P, Wolczynski S, Misrahi M. Advances in the Molecular Pathophysiology, Genetics, and Treatment of Primary Ovarian Insufficiency. Trends in endocrinology and metabolism: TEM. 2018 Jun:29(6):400-419. doi: 10.1016/j.tem.2018.03.010. Epub 2018 Apr 26
[PubMed PMID: 29706485]
Level 3 (low-level) evidence
[42]
Karacaoğlan N, Sahin U, Ercan U, Bozdogan N. One-stage repair of blepharophimosis: a new method. Plastic and reconstructive surgery. 1994 Jun:93(7):1406-9
[PubMed PMID: 8208806]
[43]
Bhattacharjee K, Bhattacharjee H, Kuri G, Shah ZT, Deori N. Single stage surgery for Blepharophimosis syndrome. Indian journal of ophthalmology. 2012 May-Jun:60(3):195-201. doi: 10.4103/0301-4738.95870. Epub
[PubMed PMID: 22569380]
[44]
Liu H, Shao Y, Zhao Z, Zhang D. One-stage correction of blepharophimosis-ptosis-epicanthus inversus syndrome using a frontalis muscle transfer technique. Journal of plastic surgery and hand surgery. 2014 Feb:48(1):74-9. doi: 10.3109/2000656X.2013.819004. Epub 2013 Aug 23
[PubMed PMID: 23968369]
[45]
Wu SY, Ma L, Tsai YJ, Kuo JZ. One-stage correction for blepharophimosis syndrome. Eye (London, England). 2008 Mar:22(3):380-8
[PubMed PMID: 17115018]
[46]
Mastellari E, La Marca A. Genetic conditions impairing female fertility. Panminerva medica. 2020 Dec:62(4):260-267. doi: 10.23736/S0031-0808.20.04208-1. Epub 2020 Nov 13
[PubMed PMID: 33185415]
[47]
Bonus ML, Pothast R, Lamb JD, Feinberg EC, Bernardi LA. Planned oocyte cryopreservation in women with blepharophimosis-ptosis-epicanthus inversus syndrome: a case series. F&S reports. 2021 Sep:2(3):332-337. doi: 10.1016/j.xfre.2021.05.006. Epub 2021 May 27
[PubMed PMID: 34553160]
Level 2 (mid-level) evidence
[48]
Meng T, Zhang W, Zhang R, Li J, Gao Y, Qin Y, Jiao X. Ovarian Reserve and ART Outcomes in Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Patients With FOXL2 Mutations. Frontiers in endocrinology. 2022:13():829153. doi: 10.3389/fendo.2022.829153. Epub 2022 Apr 28
[PubMed PMID: 35574016]
Level 2 (mid-level) evidence
[49]
Baber RJ, Panay N, Fenton A, IMS Writing Group. 2016 IMS Recommendations on women's midlife health and menopause hormone therapy. Climacteric : the journal of the International Menopause Society. 2016 Apr:19(2):109-50. doi: 10.3109/13697137.2015.1129166. Epub 2016 Feb 12
[PubMed PMID: 26872610]
[50]
Nelson LM. Clinical practice. Primary ovarian insufficiency. The New England journal of medicine. 2009 Feb 5:360(6):606-14. doi: 10.1056/NEJMcp0808697. Epub
[PubMed PMID: 19196677]
[51]
Rong WN, Ma MJ, Yang W, Yuan SQ, Sheng XL. Identification of a novel FOXL2 mutation in a fourth-generation Chinese family with blepharophimosis-ptosis-epicanthus inversus syndrome. International journal of ophthalmology. 2021:14(4):504-509. doi: 10.18240/ijo.2021.04.04. Epub 2021 Apr 18
[PubMed PMID: 33875939]
[52]
Cheng T, Yuan X, Yuan S, Zhu J, Tang S, Zhang Y. ITGB5 mutation discovered in a Chinese family with blepharophimosis-ptosis-epicanthus inversus syndrome. Open life sciences. 2021:16(1):1268-1277. doi: 10.1515/biol-2021-0129. Epub 2021 Dec 10
[PubMed PMID: 34966851]
[53]
Bashamboo A, Eozenou C, Jorgensen A, Bignon-Topalovic J, Siffroi JP, Hyon C, Tar A, Nagy P, Sólyom J, Halász Z, Paye-Jaouen A, Lambert S, Rodriguez-Buritica D, Bertalan R, Martinerie L, Rajpert-De Meyts E, Achermann JC, McElreavey K. Loss of Function of the Nuclear Receptor NR2F2, Encoding COUP-TF2, Causes Testis Development and Cardiac Defects in 46,XX Children. American journal of human genetics. 2018 Mar 1:102(3):487-493. doi: 10.1016/j.ajhg.2018.01.021. Epub 2018 Feb 22
[PubMed PMID: 29478779]
[54]
Clayton-Smith J, O'Sullivan J, Daly S, Bhaskar S, Day R, Anderson B, Voss AK, Thomas T, Biesecker LG, Smith P, Fryer A, Chandler KE, Kerr B, Tassabehji M, Lynch SA, Krajewska-Walasek M, McKee S, Smith J, Sweeney E, Mansour S, Mohammed S, Donnai D, Black G. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. American journal of human genetics. 2011 Nov 11:89(5):675-81. doi: 10.1016/j.ajhg.2011.10.008. Epub
[PubMed PMID: 22077973]
[55]
Oley C, Baraitser M. Blepharophimosis, ptosis, epicanthus inversus syndrome (BPES syndrome). Journal of medical genetics. 1988 Jan:25(1):47-51
[PubMed PMID: 3270326]
[58]
Saint-Laurent C, Mazeyrie L, Yart A, Edouard T. Novel therapeutic perspectives in Noonan syndrome and RASopathies. European journal of pediatrics. 2024 Mar:183(3):1011-1019. doi: 10.1007/s00431-023-05263-y. Epub 2023 Oct 21
[PubMed PMID: 37863846]
Level 3 (low-level) evidence
[59]
Singh N, Verma P, Bains R, Mutalikdesai J. Apert syndrome: craniofacial challenges and clinical implications. BMJ case reports. 2024 Jul 16:17(7):. pii: e260724. doi: 10.1136/bcr-2024-260724. Epub 2024 Jul 16
[PubMed PMID: 39013624]
Level 3 (low-level) evidence
[60]
Nuovo S, Passeri M, Di Benedetto E, Calanchini M, Meldolesi I, Di Giacomo MC, Petruzzi D, Piemontese MR, Zelante L, Sangiuolo F, Novelli G, Fabbri A, Brancati F. Characterization of endocrine features and genotype-phenotypes correlations in blepharophimosis-ptosis-epicanthus inversus syndrome type 1. Journal of endocrinological investigation. 2016 Feb:39(2):227-33. doi: 10.1007/s40618-015-0334-3. Epub 2015 Jun 23
[PubMed PMID: 26100530]
[61]
Amer AA, Abdellah MM, Hassan NHF, Mounir A. Surgical outcome of epicanthus and telecanthus correction by C-U medial canthoplasty with lateral canthoplasty in treatment of Blepharophimosis syndrome. BMC ophthalmology. 2022 May 19:22(1):226. doi: 10.1186/s12886-022-02455-2. Epub 2022 May 19
[PubMed PMID: 35590300]
[62]
Sa HS, Lee JH, Woo KI, Kim YD. A new method of medial epicanthoplasty for patients with blepharophimosis-ptosis-epicanthus inversus syndrome. Ophthalmology. 2012 Nov:119(11):2402-7. doi: 10.1016/j.ophtha.2012.05.037. Epub 2012 Jul 24
[PubMed PMID: 22835816]
[63]
Savino G, Mandarà E, Calandriello L, Dickmann A, Petroni S. A Modified One-Stage Early Correction of Blepharophimosis Syndrome Using Tutopatch Slings. Orbit (Amsterdam, Netherlands). 2015:34(4):186-91. doi: 10.3109/01676830.2015.1015146. Epub 2015 Jun 4
[PubMed PMID: 26043072]
[64]
Fokstuen S, Antonarakis SE, Blouin JL. FOXL2-mutations in blepharophimosis-ptosis-epicanthus inversus syndrome (BPES); challenges for genetic counseling in female patients. American journal of medical genetics. Part A. 2003 Mar 1:117A(2):143-6
[PubMed PMID: 12567411]