Continuing Education Activity
Tricuspid atresia is a congenital heart defect that results in cyanosis due to the absence of communication between the right atrium and ventricle caused by the complete agenesis of the tricuspid valve. This condition has several subtypes with varied clinical presentations determined by the extent of pulmonary blood flow. The root cause of this condition lies in abnormal heart development during embryogenesis without any confirmed genetic predisposition. Without intervention within the first year of life, tricuspid atresia carries a high mortality rate.
The anatomical variations in tricuspid atresia, often involving pulmonary obstruction, prompt a thorough evaluation to assess its impact on hemodynamic status. The lesion is further classified into several subtypes, with or without pulmonary obstruction, based on the anatomical changes. Implementing further procedural recommendations based on the evaluation aims to restore normal blood flow. Given the potential for long-term complications, intensive care serves as the primary setting for managing tricuspid atresia, followed by ongoing monitoring to ensure a sustained high quality of life for affected individuals. This activity describes the etiology, pathophysiology, diagnosis, complications, and management of tricuspid atresia. This activity equips healthcare professionals with updated knowledge and skills for effective diagnosis, treatment, and overall management of this congenital heart condition, leading to improved patient care and outcomes.
Objectives:
Identify clinical manifestations and anatomical variations associated with tricuspid atresia, enabling prompt diagnosis.
Apply evidence-based interventions and procedural recommendations for restoring normal blood flow in patients with tricuspid atresia.
Assess the impact of anatomical variations and pulmonary obstruction on hemodynamic status, guiding comprehensive patient assessment.
Coordinate care plans and interventions for patients with tricuspid atresia, emphasizing a patient-centered approach and continuous follow-up.
Introduction
Tricuspid atresia is a cyanotic congenital heart defect characterized by the complete agenesis of the tricuspid valve. Consequently, the absence of communication between the right atrium and the ventricle leads to cyanosis. The condition has several subtypes with varied clinical presentations determined by the extent of pulmonary blood flow.[1] The root cause of this condition lies in abnormal heart development during embryogenesis without any confirmed genetic predisposition. Without intervention within the first year of life, tricuspid atresia carries a high mortality rate.
The anatomical variations in tricuspid atresia, often involving pulmonary obstruction, prompt a thorough evaluation to assess the degree of the lesion and its impact on a patient's hemodynamic status. The lesion is further classified into several subtypes, with or without pulmonary obstruction, based on the anatomical changes. Implementing further procedural recommendations based on the evaluation aims to restore normal blood flow. Given the potential for long-term complications, intensive care serves as the primary setting for managing tricuspid atresia, followed by ongoing monitoring to ensure a sustained high quality of life for affected individuals.
Etiology
The pathogenesis of tricuspid atresia is not fully understood, but it occurs due to the disruption of the normal development of the atrioventricular valves from the endocardial cushion. In most patients, the tricuspid inlet manifests as a dimple in the right atrium, presenting in a muscular form. In less common occurrences, a fusion involves partially delaminated leaflets, resulting in the formation of membranes, as observed in the Ebstein type.[2]
Epidemiology
The overall prevalence of congenital heart disease is 81 per 10,000 births. Tricuspid atresia ranks as the third most common cyanotic congenital heart disease, with an occurrence of approximately 1.2 per 10,000 live births. The condition shows no gender preference, affecting males and females equally.[3][4] Notably, there is no significant familial recurrence risk associated with tricuspid atresia, and instances of such recurrence are rare in the literature. When familial cases do occur, they are believed to follow an autosomal recessive inheritance pattern.[5]
Pathophysiology
Blood flows back to the heart through the superior and inferior vena cava, entering the right atrium. However, the tricuspid valve prevents blood from passing through, as it is affected by atresia, resulting in a hypoplastic right ventricle.
An obligatory right-to-left shunt develops at the atrial level due to impeded forward blood flow across the tricuspid valve. This leads to the mixing of systemic and pulmonary venous blood in the left atrium. The amount of oxygen-saturated blood reaching the left ventricle, aorta, and, subsequently, the entire body is contingent on the relative volumes of pulmonary and systemic venous return.
The amount of pulmonary blood flow is determined by the degree of pulmonary obstruction, the presence of ventricular septal defect (VSD), and the relationship of the great arteries.
Patients with pulmonary obstruction due to pulmonary stenosis (PS) or atresia exhibit reduced pulmonary venous return, resulting in diminished systemic arterial oxygen saturation and the development of cyanosis. In contrast, individuals without pulmonary obstruction tend to have increased pulmonary venous return, leading to relatively higher systemic arterial saturation and a lower likelihood of cyanosis.
Classification of Tricuspid Atresia
The classification system for tricuspid atresia offers a comprehensive framework for categorizing a specific cardiac lesion based on key factors, including the relationship of the great arteries, the presence of a VSD, and the degree of pulmonary obstruction.[1][6][7] The lesion is further detailed under 3 main types, comprising distinct subgroups delineating variations in anatomical characteristics and associated conditions.
Type I (70% to 80%): This type is characterized by normal anatomy of the great arteries and is further divided into subgroups.
- Subgroup a: Intact ventricular septum with pulmonary atresia
- Subgroup b: Small VSD with PS or hypoplasia
- Subgroup c: Large VSD without PS
Type II (12% to 25%): This type involves D-transposition of the great arteries (D-TGA) and is further divided into subgroups.
- Subgroup a: VSD with pulmonary atresia
- Subgroup b: VSD with PS or hypoplasia
- Subgroup c: VSD without PS
Type III (3% to 6%): This type encompasses malposition defects, such as truncus arteriosus, atrioventricular septal defects, and double outlet right ventricle, involving the great arteries other than D-TGA.
Blood Flow in Patients With Pulmonary Obstruction
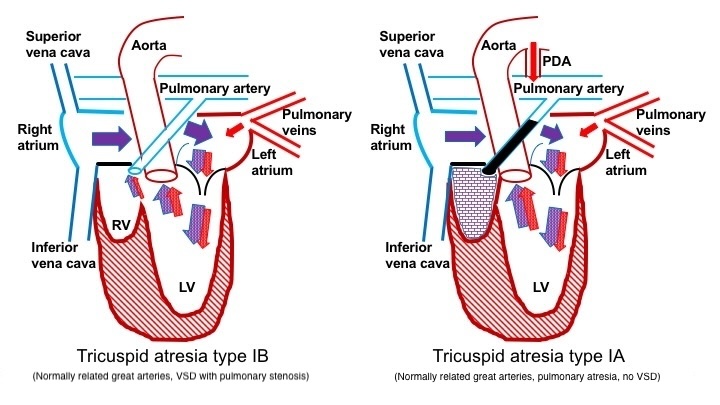
Pulmonary atresia develops in patients with normally related great arteries and no VSD. Hence, the blood supply to the lungs depends on a patent ductus arteriosus (PDA). In type Ia tricuspid atresia, blood is redirected from the right atrium across the atrial septum to the left atrium, passing through the mitral valve into the left ventricle. Subsequently, blood exits the heart through the aorta to supply blood to the body and concurrently travels through the PDA to reach the lungs. In the presence of a VSD, blood flows through the VSD into the pulmonary arteries. The degree of pulmonary blood flow would correlate with the size of the VSD and the extent of PS in types Ib and Ic tricuspid atresia (see Image. Blood Flow in Patients With Pulmonary Obstruction).
Blood Flow in Patients Without Pulmonary Obstruction
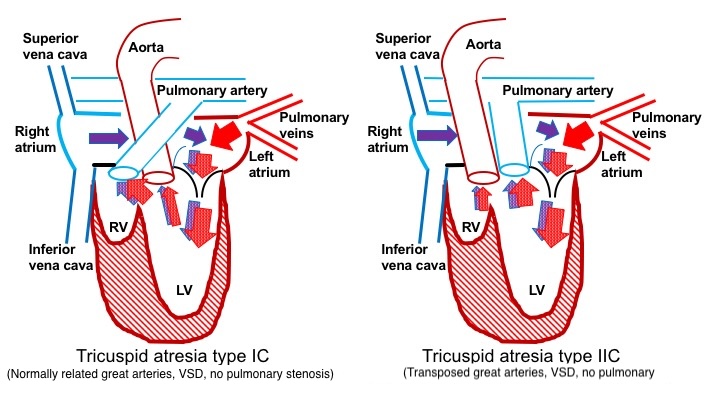
Patients with transposition of the great arteries typically exhibit VSDs and commonly have an unobstructed pulmonary blood supply (type IIc). Unlike other types, these patients do not depend on the PDA for pulmonary blood flow. In type II tricuspid atresia, blood from the right atrium flows into the left atrium through the mitral valve into the left ventricle and then exits through the pulmonary artery (see Image. Blood Flow in Patients Without Pulmonary Obstruction).
Furthermore, blood flows across the VSD into the aorta, supplying blood to the body. In tricuspid atresia types IIa and IIb, some degree of pulmonary obstruction may exist, which may require using a PDA for pulmonary blood flow. In types IIa and IIb, blood flows from the right atrium into the left atrium, across the mitral valve, into the left ventricle, through the VSD into the aorta, and finally, via the PDA, reaching the lungs.
History and Physical
As mentioned above, the clinical presentation is contingent upon the degree of pulmonary obstruction, the presence of a VSD, and the relationship of the great arteries.
Patients with Pulmonary Obstruction
Patients often present with cyanosis in the newborn period, especially after the closure of the ductus. Findings from physical examination reveal central cyanosis with normal pulses and potentially a diminution in the right ventricular impulse due to tricuspid atresia with pulmonary oligemia. The presence of a thrill may indicate a restrictive VSD or severe PS. A holosystolic murmur at the left lower sternal border indicates a VSD; at times, a continuous murmur from the PDA may be auscultated. A systolic ejection murmur audible at the left upper sternal border corresponds to PS. Clubbing may manifest in older patients with unrepaired tricuspid atresia and chronic cyanosis.
Patients without Pulmonary Obstruction
Patients with a VSD and without PS often experience substantial pulmonary blood flow. Such cases could be missed at birth, as these individuals do not present with cyanosis. The condition may become apparent when pulmonary vascular resistance drops, leading to pulmonary overcirculation. This condition manifests with signs and symptoms of heart failure, such as tachypnea or respiratory distress, poor feeding, and impaired growth. Notable findings upon physical examination include tachypnea, tachycardia, and hepatomegaly in patients exhibiting pulmonary plethora.[8]
Evaluation
With the advancements in antenatal ultrasound and fetal echocardiogram programs, most patients are now diagnosed antenatally in the United States. The mean gestational age at diagnosis is around 22 weeks, although it can be as early as 11 weeks.[9][10][11][12][13] According to a recent large multicenter study, the prevalence of prenatally diagnosed tricuspid atresia ranges between 0.2 and 0.9 per 10,000, with an observed increase in the proportion of cases diagnosed over time.[14]
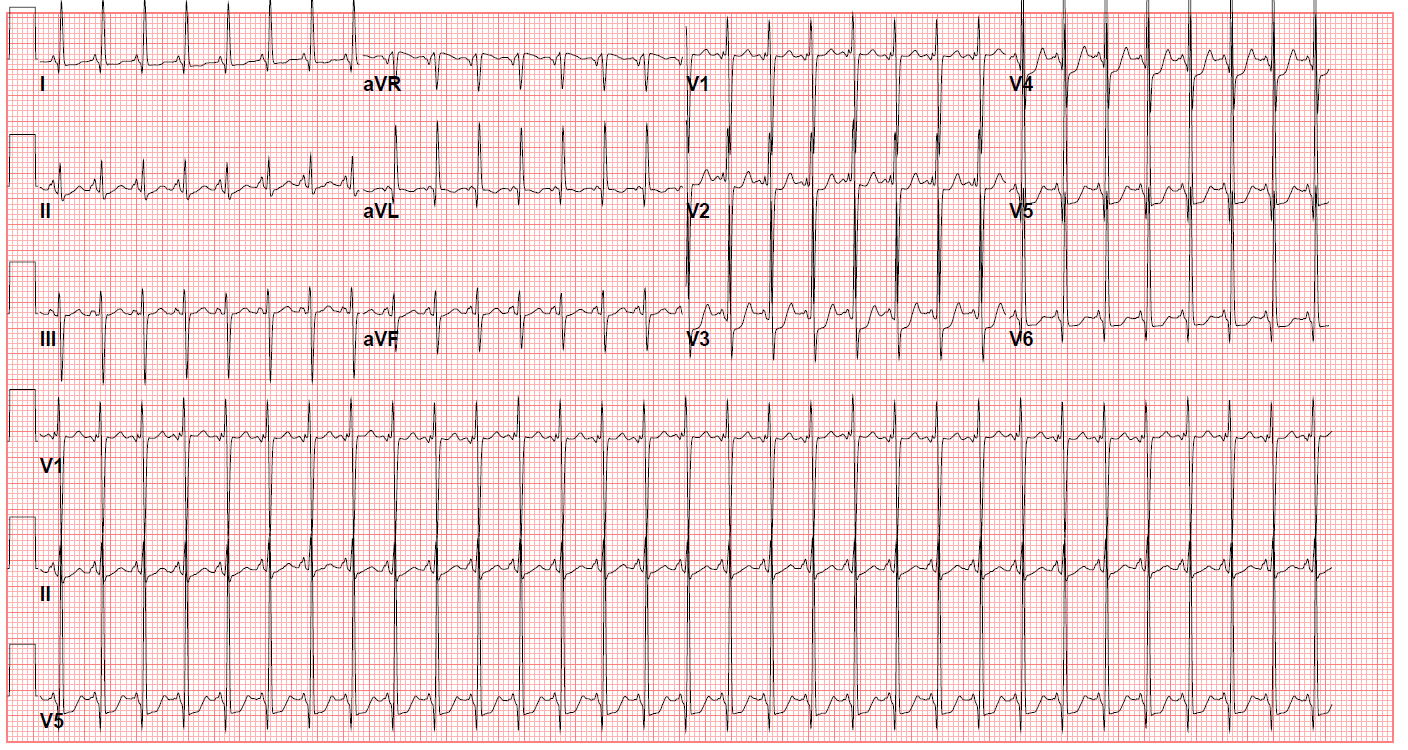
Clinicians may identify patients not diagnosed antenatally at the time of critical congenital heart disease screening with hypoxemia or the presence of a heart murmur. A chest x-ray may show decreased pulmonary vascular markings, which signify pulmonary oligemia. A prominent right heart border may also indicate right atrial dilation. The electrocardiogram (ECG) of the lesion is characterized by a left superior QRS axis (-30 to -90), representing a characteristic finding (see Image. ECG of a Patient With Tricuspid Atresia). Diminished right ventricular forces and potential evidence of left ventricular hypertrophy may also be observed.[15]
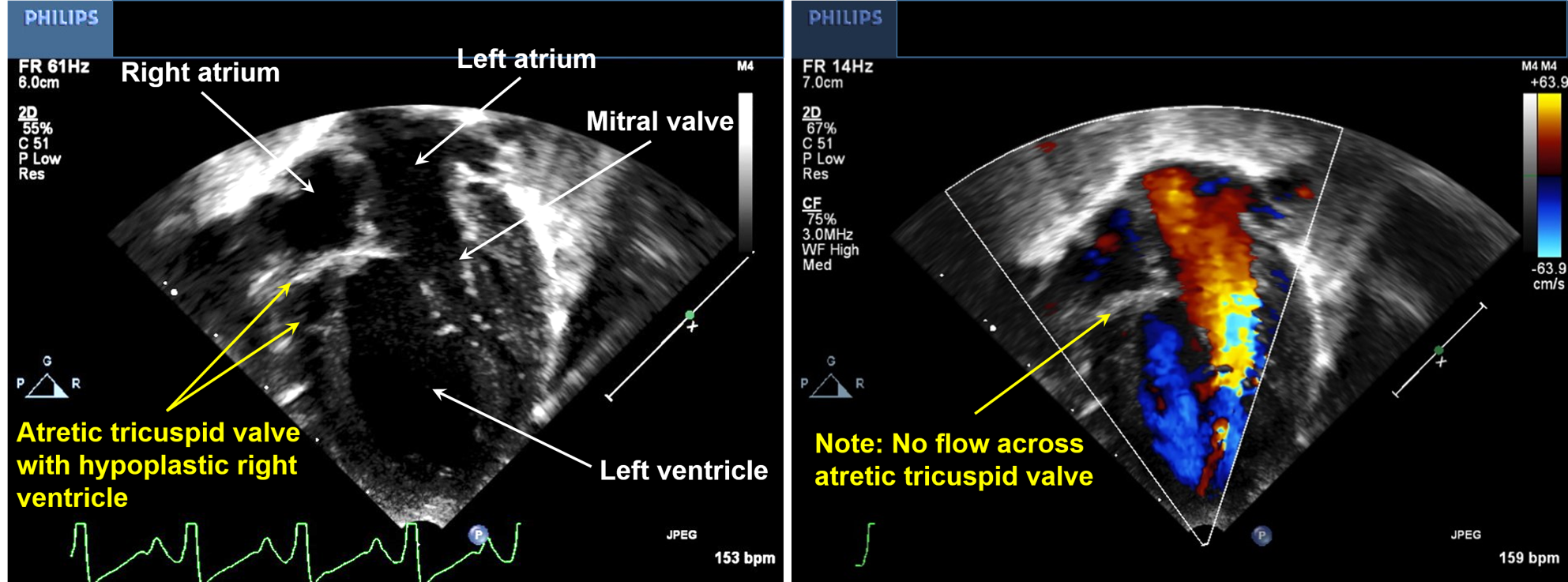
Echocardiography serves as a diagnostic tool for tricuspid atresia. Two-dimensional echocardiography would show the absence of the tricuspid valve and disproportionate sizes of the ventricular cavities, with the left ventricle larger than the right. Color flow Doppler demonstrates the lack of flow across the tricuspid valve (see Image. Echocardiography of a Patient With Tricuspid Atresia).
Cardiac catheterization is not usually necessary for diagnostic purposes in this condition. However, in cases where the atrial septum communication is restrictive and impedes sufficient blood flow from the right atrium to the left atrium, a cardiac catheterization procedure, specifically a balloon atrial septostomy, may become necessary. Genetic testing may be considered in this condition due to its associations with trisomies, VACTERL syndrome, and 22q11.[16][17][10]
Treatment / Management
The initial management of the condition focuses on stabilizing the patient. The initiation of prostaglandin soon after birth is imperative in cyanotic patients with severe or critical PS or a very small VSD, relying on a ductal-dependent pulmonary blood supply. Patients presenting later in infancy with heart failure and pulmonary over-circulation may require diuretics as part of the indicated treatment.
Due to the presence of only one functional ventricle (the left ventricle), individuals with tricuspid atresia undergo staged single ventricle palliation to ensure sufficient pulmonary and systemic blood flow, ultimately leading to the separation of these circulatory systems. The selection of the surgical approach for the initial stage of palliation depends on factors such as the anatomy of the great vessels, the presence or absence of outflow tract obstruction, and the size of any existing VSD.
Patients With Pulmonary Obstruction
For patients with pulmonary obstruction, the initial stage involves ensuring sufficient pulmonary blood flow through a systemic-to-pulmonary artery shunt, commonly achieved with a modified Blalock-Taussig (BT) shunt, also referred to as a BT-Thomas shunt. This involves creating a connection between the right subclavian and pulmonary artery, typically utilizing a polytetrafluoroethylene tubular graft.
Patients Without Pulmonary Obstruction
For patients with unobstructed pulmonary blood flow (type IC), the initial stage may involve the placement of a pulmonary artery (PA) band to regulate excessive blood flow to the pulmonary vascular bed. In a subset of cases, the VSD may be sufficiently small to naturally limit pulmonary blood flow, resulting in a balanced circulation. Therefore, pulmonary artery banding is unnecessary for these patients as they have balanced circulation and proceed to the second stage of palliation, as detailed below.
Patients With D-TGA and Subaortic Obstruction
For patients with type II lesions characterized by a VSD that obstructs aortic blood flow, the first stage may involve enlarging the VSD or implementing a Damus-Kaye-Stansel procedure (an anastomosis between the main pulmonary artery and ascending aorta) in conjunction with a modified BT shunt.
Second and Third Stages
The second stage for type I and II lesions involves cavo-pulmonary anastomosis, where the superior vena cava connects to the right pulmonary artery, achieved through either the bidirectional Glenn or hemi-Fontan procedure. This modification facilitates passive blood flow from the upper body into the pulmonary vessels. This surgery is typically performed at around 6 months.
The third stage is the Fontan procedure, which was first described in 1971 for tricuspid atresia.[18] The original Fontan involved end-to-end anastomosis between the right atrial appendage and the proximal end of the right pulmonary artery, but the procedure has evolved significantly since then. In the current era, it involves either an extra-cardiac or intra-atrial non-valved conduit connecting the inferior vena cava to the pulmonary arteries, and this conduit may be fenestrated. Fenestration serves as a "pop-off" valve during the adjustment of the lungs to the increased blood flow from the lower body.[19] The Fontan procedure directs total systemic venous return passively into the pulmonary vessels and is typically performed when the patient is between the ages of 2 and 3. Patients experiencing progressive ventricular dysfunction may eventually necessitate heart transplantation.[20]
Differential Diagnosis
The differential diagnoses for this condition include other lesions characterized by cyanosis and reduced pulmonary blood flow, including isolated pulmonary atresia and tetralogy of Fallot, as well as atrial septal defect, pulmonic stenosis, pulmonary atresia, and tricuspid stenosis.
Prognosis
The majority of unoperated patients face a high risk of mortality within the first year of life.[21] In the current era of early diagnosis and effective surgical interventions, individuals with tricuspid atresia commonly survive well into adulthood, maintaining good functional capacity. Operative mortality associated with the Fontan procedure is consistently below 2%. A comprehensive study conducted by Sittiwangkul et al in 2004, focusing specifically on outcomes in tricuspid atresia patients (between 1971 and 1999), revealed survival rates of 82%, 72%, and 61% at 1, 5, and 20 years, respectively.[22]
Surgical outcomes have significantly improved due to advancements in surgical techniques and innovations for addressing patients' conditions. A recent study conducted by Mery et al, examining outcomes of patients undergoing the Fontan procedure for various pathologies, including tricuspid atresia, demonstrated a transplant-free survival rate of 92% at 15 years and an 87% freedom from Fontan failure.[23]
Complications
Each stage of palliation in tricuspid atresia comes with numerous short and long-term complications. The period between the first and second stages of surgical palliation for single ventricles exhibits the highest rate of interstage mortality. Patients who have undergone BT shunts face various complications, including the risk of shunt obstruction. In the current era, the mortality rate remains elevated among patients who have undergone a modified BT shunt. A recent study examining outcomes in patients after a BT shunt revealed an in-hospital mortality rate of 12%, accompanied by an additional 6% interval mortality. BT shunt-related thrombosis and stenosis occurred in 20% of patients, and non-cardiac complications, such as necrotizing enterocolitis and cerebral vascular accidents, were notable occurrences after BT shunts.[24] Currently, most single ventricle palliation programs incorporate home monitoring initiatives for shunted patients, which have proven successful in enhancing outcomes for this patient population.[25]
Complications encountered after pulmonary artery band placement include band migration, with the potential need for re-intervention if the band is too loose. Research has also documented instances of pulmonary artery stenosis or distortion. A recent study highlighted that utilizing bilateral PA bands, particularly those with smaller diameter bands and longer placement, necessitates numerous pulmonary arterial interventions.[26]
The Glenn surgery has excellent short and long-term outcomes, with operative mortality of less than 1% and 5-year survival of 87%.[27] Although long-term complications are infrequent after this surgery, thrombosis and thromboembolic events present with the highest associated mortality.[28] In addition, long-term complications associated with Fontan include arrhythmias, ventricular dysfunction, cyanosis, and protein-losing enteropathy (PLE).
Systolic and diastolic ventricular dysfunction emerges as a long-term complication following single ventricle palliation. In the neonatal phase after the Blalock-Taussig (BT) shunt, the single effective ventricle undergoes increased volume load, potentially leading to valvular regurgitation and heightened hemodynamic stress on the myocardium. Chronic hypoxia in the context of intracardiac mixing further challenges the myocardium. Following the Fontan procedure, the ventricle transitions from volume-overloaded to volume-depleted, causing acute systolic and diastolic dysfunction in the short term. However, the long-term impact involves altered volume load and ventricular mass ratios, potentially influencing ventricular mechanics over time.[29] Systolic ventricular dysfunction stands as an independent risk factor for chronic Fontan failure.[23] In cases of failed Fontan circulation, transplantation may become a necessary intervention.[30][20]
Arrhythmias are common after the Fontan procedure, with atrial tachycardias being the predominant type, often attributed to suture lines in the right atrium that may interfere with atrial conduction. The incidence of this complication has decreased with the extracardiac modification of surgery.[31][23] Although ventricular tachyarrhythmias have been reported in these patients, they are notably less common than atrial arrhythmias.
Thromboembolism is a potential concern in these patients, with an overall prevalence of approximately 11%. It may manifest in various locations, including the Fontan conduit, right atrium, lungs (pulmonary embolism), or brain (stroke-systemic arterial emboli). The risk of thromboembolism is heightened in the presence of an atriopulmonary connection or atrial arrhythmias.[32] A pervasive debate surrounds the use of antiplatelet therapy versus anticoagulation to mitigate this risk. Data indicate that the rates of thromboembolic and bleeding events associated with antiplatelet therapy are comparable to those linked with anticoagulation therapy.[33] Recent American Heart Association guidelines propose that it is reasonable for all patients with Fontan circulation to receive antiplatelet therapy. Anticoagulation should be reserved for those with presumed risk factors, such as arrhythmias or previous thrombosis.[34] Ongoing research is exploring newer direct oral anticoagulants in pediatric cardiac populations, offering a potential future alternative to warfarin.
PLE is a well-documented complication and occurs in 5% to 12% of patients after Fontan.[35] This condition arises from lymphatic congestion, leading to the leakage of lymph-rich material into the low-pressure intestinal tract. Longstanding PLE can affect the nutritional status of the patient. Supportive treatment is the mainstay for PLE and involves strategies such as decreasing fluid overload with diuretic therapy, replacing albumin and immunoglobulin, and adopting a high-protein, low-fat diet. Enteral corticosteroid use has demonstrated maintenance of albumin levels and decreased symptoms.[34]
In addition, plastic bronchitis is another possible long-term sequelae in patients with Fontan circulation. Thick secretions in the airway lumen characterize this condition and occur in around 4% of patients. Plastic bronchitis is believed to be caused by spillage of proteinaceous lymph through lymphatic-bronchial communications.[36] The variability in lymphatic anatomy may explain why only certain patients with similar degrees of venous hypertension develop PLE or plastic bronchitis while others do not.
Patients who have undergone the Fontan procedure may experience cyanosis due to various factors. Right-to-left shunting can occur across the fenestration in a Fontan. Systemic and pulmonary venous collaterals may develop, resulting in right-to-left shunting from the Fontan pathway to the atrium. Cyanosis may be exacerbated during exercise. In some cases, patients may find relief through interventions such as transcatheter fenestration closure or venovenous occlusion.[34]
Tricuspid atresia can lead to various long-term complications, including Fontan-associated liver disease (characterized by liver fibrosis and cirrhosis), renal dysfunction, somatic growth problems, psychosocial and neurodevelopmental issues, and decreased exercise tolerance.
Deterrence and Patient Education
Patients with tricuspid atresia necessitate consistent and lifelong monitoring by a cardiologist specialized in congenital heart disease, such as a pediatric cardiologist or adult congenital heart disease (ACHD) specialist. The frequency of follow-up appointments varies across stages of single ventricle palliation, with those who have undergone the Fontan procedure typically requiring at least yearly follow-up.
History and physical examination are beneficial in predicting many of the complications described above. Although mild jugular venous distension is typical following cavo-pulmonary anastomosis, marked jugular venous distension and hepatomegaly warrant additional assessment for potential obstruction in Glenn or Fontan procedures or systolic ventricular dysfunction of the heart. Edema and diarrhea may be present in the context of PLE. Palpitations in the medical history should trigger additional assessment for arrhythmias. Respiratory distress may indicate heart failure, while a history of thick airway secretions may suggest the presence of plastic bronchitis. A comprehensive history and physical examination during follow-up visits are essential for monitoring these patients effectively.
In addition to the clinical assessment during yearly follow-ups, an annual ECG and transthoracic echocardiogram are necessary. A more comprehensive cardiovascular and extracardiac evaluation should be conducted every 3 to 4 years to monitor for the aforementioned complications. The comprehensive cardiovascular surveillance may involve a 24-hour Holter monitor, exercise stress test, pro-brain natriuretic peptide measurement, cardiac magnetic resonance imaging, computed tomography angiography, and cardiac catheterization. Organ system surveillance includes monitoring organ function through blood tests and extracardiac imaging.[34][37]
Patients with surgically corrected tricuspid atresia can survive into childbearing age. Pregnancy has inconclusive effects on patients with Fontan circulation. Nonetheless, such pregnancies are considered high-risk due to increased susceptibility to arrhythmias, heart failure, and thromboembolic complications. Counseling should encompass overall cardiovascular health and the condition of noncardiac organ systems. These patients require collaborative care and treatment of their condition by an ACHD specialist and obstetrician specializing in high-risk pregnancies or a maternal-fetal medicine specialist.[34][38]
Pearls and Other Issues
Exploring crucial insights and noteworthy considerations, this section delves into other significant aspects related to tricuspid atresia, including diagnostic clues, long-term outcomes, and historical milestones in Fontan palliation.
- A high index of suspicion is necessary for tricuspid atresia in newborns with cyanosis and left axis deviation or left ventricular hypertrophy on ECG.
- Patients with tricuspid atresia have a single left ventricle (systemic ventricle), resulting in the most favorable long-term outcomes among single ventricle cases undergoing the Fontan procedure.
- Among single ventricles, tricuspid atresia was the first type for which the Fontan palliation was performed in 1971.
Enhancing Healthcare Team Outcomes
An interprofessional team approach is imperative for treating patients with tricuspid atresia. Patients diagnosed with tricuspid atresia are typically cared for in a neonatal intensive care unit (ICU) before and after surgery in a cardiac ICU. A collaborative approach among pediatricians, intensivists, cardiologists, and surgeons is crucial for favorable outcomes. Given the potential involvement of non-cardiac organ systems in some patients due to associated syndromes, consultation with other pediatric sub-specialists becomes necessary. Nurses, respiratory therapists, perfusionists, nutritionists, and social workers are integral to a comprehensive interprofessional medical team.[39] Critical care and neonatal nurses, in particular, are instrumental in monitoring patients, documenting their status, and ensuring effective communication across the entire team.
Upon discharge, patients with tricuspid atresia require a medical home and comprehensive care due to the complexity of their condition. In any debate regarding anticoagulation versus platelet-blocking, the involvement of a board-certified cardiology pharmacist is crucial. Their responsibilities include performing medication reconciliation, verifying the dosages of all drugs, and counseling patients, their families, and team members. A primary care clinician is essential for coordinating care and minimizing the burden on the family.[40]
Numerous studies are investigating neurodevelopmental outcomes in the cohort of patients with tricuspid atresia, who are prone to delayed development, attention-deficit hyperactivity disorder, and learning disorders.[41][42] As individuals with tricuspid atresia are now experiencing longer lifespans, the role of the ACHD specialist becomes increasingly critical in providing care and monitoring for potential long-term complications. The 2018 guidelines for treating adults with congenital heart disease emphasize the need for a smooth and effective transition of care from pediatric cardiologists to ACHD specialists, particularly during the transition from late adolescence to early adulthood.[37] This collaborative interprofessional team paradigm is anticipated to yield superior outcomes.