Continuing Education Activity
A supernumerary digit or polydactyly is a frequently occurring congenital malformation, predominantly affecting the upper limbs. Individuals affected by this condition typically exhibit excessive fingers or toes, exceeding the usual count of 5. Polydactyly can be categorized into 3 distinct classifications—preaxial, postaxial, and central or mesoaxial—each based on the specific anatomical location involved. Although supernumerary digits typically manifest as isolated occurrences, they can also be associated with specific syndromes. This activity reviews the etiology, pathophysiology, evaluation, and surgical interventions related to supernumerary digits. This topic also highlights the significance of the interprofessional healthcare team in identifying syndromic associations or potential involvement of other organs by conducting comprehensive evaluations to assess for underlying conditions.
Objectives:
Identify the presence of supernumerary digits through comprehensive clinical evaluation, including physical examination and imaging techniques.
Implement evidence-based evaluation and diagnostic procedures to determine the extent and categorization of supernumerary digits, which can involve preaxial, postaxial, or central locations.
Apply appropriate treatment modalities for supernumerary digits, including surgical intervention or non-surgical management, based on the specific case and patient's needs.
Collaborate with specialists from an interprofessional healthcare team, including orthopedics, genetics, plastic surgeons, pediatrics, and occupational and physical therapists, to ensure comprehensive care for patients with supernumerary digits.
Introduction
A supernumerary digit, or polydactyly, is a congenital anomaly affecting the upper or lower extremities and is typically identified immediately after birth.[1] Polydactyly is the most common congenital anomaly of the hand and foot, where affected individuals typically exhibit excessive fingers or toes, exceeding the usual count of 5.[1] Parey, in the sixteenth century, documented individuals with more than the standard count of 5 fingers as having "superfluous fingers."[1]
Apart from Parey's historical description, artwork discovered in the southwestern "Four Corners" region of the United States portrays individuals with 6 digits on their hands and feet. This artwork is believed to have its origins in an early population that existed between AD 600 and 1280.[1]
The classification of polydactyly is contingent upon the location of the supernumerary digit. Preaxial polydactyly involves the radial or great toe side, postaxial polydactyly relates to the ulnar or fifth toe side, and central or mesoaxial polydactyly encompasses the second through fourth digits.[1] Polydactyly is primarily an autosomal dominant anomaly and may manifest as an isolated condition or be associated with a syndrome. A comprehensive physical examination of newborns with polydactyly is imperative to evaluate the potential presence of associated syndromic disorders. This topic discusses the etiology and pathophysiology of polydactyly, as well as the significance of the interprofessional team in evaluating and treating affected patients.
Etiology
Polydactyly is primarily an autosomal dominant anomaly that exhibits variable penetrance. Polydactyly results from a disruption in digit control during development.[1] Currently, a minimum of 10 genetic loci and 6 genes are recognized as contributors to the non-syndromic form of polydactyly. Limb and digit development is guided by multiple signal centers between 4 and 8 weeks of gestation. Sequence variants in the sonic hedgehog (SHH) enhancer ZRS (zone of polarizing activity regulatory sequence) and GLI3 (glioma-associated oncogene homolog 3) cause at least 6 different forms of polydactyly. Variants in the SHH gene are responsible for thumb or preaxial polydactyly type I, the most prevalent form of this condition. Expression of SHH within a region known as the zone of polarizing activity (ZPA) is regulated by the expression of the ZRS and is responsible for the development of the anterior-posterior (radial-ulnar) axis of the limb.[2]
SHH is crucial for the development of digit numbers and types.[1] In typical hand development, the length of the limb is determined by signals from the fibroblast growth factor originating from the apical ectodermal ridge. Digit development unfolds along a radial-ulnar axis and is controlled by bone morphogenetic proteins (BMP) family members in conjunction with SHH.[3] SHH is present in higher concentrations on the ulnar side compared to the radial side, contributing to the differentiation of digits.[4] Mutations in the LMBR1 gene have been associated with the onset of polydactyly due to upregulated SHH expression during limb development.[5]
Preaxial polydactyly type IV and postaxial polydactyly types A1 and B are associated with heterozygous mutations in the GLI3 gene. In a subset of individuals with Greig cephalopolysyndactyly, there is a missense mutation in the GLI3 gene, which functions as an antagonist to SHH. This missense mutation can result in an upregulation of SHH, potentially leading to polydactyly by disrupting the establishment of the radial-ulnar axis.[2][6]
Epidemiology
Polydactyly is estimated to manifest in approximately 1.6 to 10.7 cases per 1000 live births, with males being affected twice as frequently as females.[7] The incidence of polydactyly of the fingers is higher in Blacks, which is approximately 1 in 300 individuals, than in Whites, which is around 1 in 3000 individuals. Preaxial polydactyly is more prevalent in White, American Indian, and Asian populations, whereas postaxial polydactyly is more frequently observed in Black populations.[8] The prevalence of postaxial polydactyly is estimated to be between 1 and 2 cases per 1000 live births.[7] In contrast, preaxial polydactyly is less common, with reported incidences ranging from 0.08 to 1.4 cases per 1000 to 1 in 3000 live births.[8][9]
Pathophysiology
In embryonic limb development, the typical pattern of digit formation is orchestrated through intricate interactions among multiple signaling molecules. Supernumerary digits can occur on one or both hands and feet when there is an upregulation or downregulation in the expression of these signaling molecules. This upregulation or downregulation often results from genetic errors in various chromosomal regions. Most commonly, polydactyly of the hand occurs, with a greater involvement of the ulnar side than the radial side.[7]
History and Physical
As a congenital disorder, polydactyly can exhibit familial patterns. Thus, obtaining an accurate family history is crucial for assessment and diagnosis. Patients with polydactyly typically present with an extra finger or toe, which is commonly detected in the immediate postnatal period. Findings from the physical examination include the identification of a supernumerary digit. Imaging, such as radiographs, is valuable for surgical planning and, more precisely, for characterizing the specific polydactyly pattern.
Preaxial (Radial) Polydactyly of the Hand
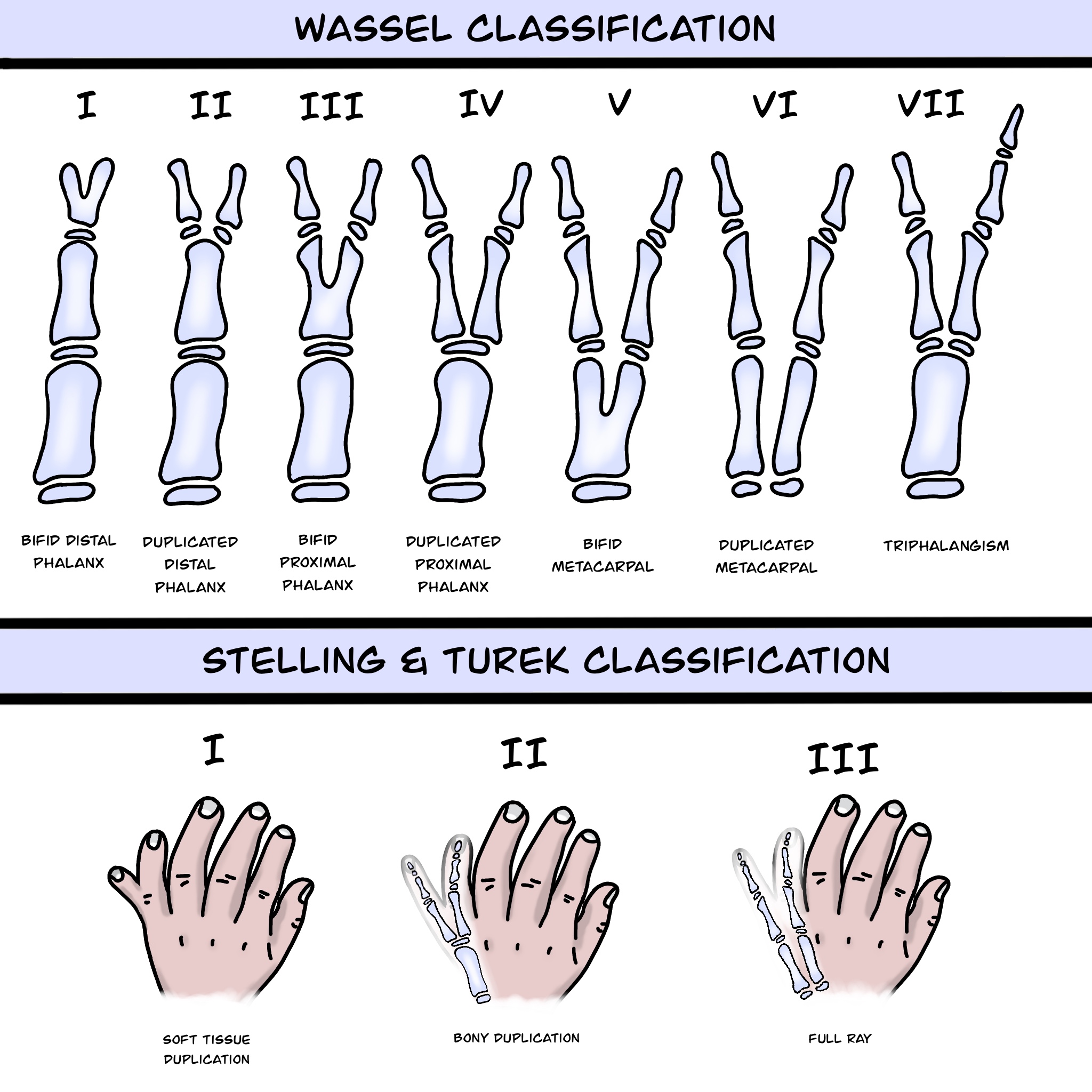
Preaxial polydactyly of the hand is characterized by an additional digit growing on the radial side of the hand. The Wassel classification system is used to classify the preaxial polydactyly of the hand into 7 classes, as mentioned below (see Image. Polydactyly Classifications).
- Type I involves a bifid distal phalanx.
- Type II involves a duplicated distal phalanx.
- Type III involves bifid proximal phalanges.
- Type IV involves a duplicated proximal phalanx.
- Type V involves a bifid metacarpal.
- Type VI involves duplicated metacarpals.
- Type VII consists of triphalangia.[10]
Postaxial Polydactyly
Postaxial polydactyly entails the duplication of the small finger along the ulnar border of the hand or the lateral border of the foot. The Stelling and Turek classification system describes 3 types of postaxial polydactyly of the hand, as mentioned below (see Image. Postaxial Polydactyly).
- Type 1 involves soft tissue duplication.
- Type 2 has bony duplication.
- Type 3 includes a full ray, including the metacarpal.[11]
Central Polydactyly
Central polydactyly, which involves the duplication of the index, middle, or ring fingers or the second, third, or fourth toes, represents a considerably smaller proportion of polydactyly cases.[11] The ring finger is more commonly affected.[8] Central polydactyly is also primarily an autosomal dominant inherited disorder associated with syndactyly or polydactyly of the feet.[8]
Central polydactyly is classified into 3 distinct types, each defined by specific anatomical characteristics, as mentioned below.
- Type 1 involves a duplication with no adjacent finger attachments.
- Type 2 exhibits shared joints, a bifid metacarpal, or phalanx with an adjacent finger.
- Type 3 features a fully formed independent digit, including the metacarpal.[11]
Foot Polydactyly
The Venn-Watson classification is the primary system employed to categorize polydactyly of the foot, featuring 6 subdivisions based on the metatarsal anatomy, as mentioned below.
- Class 1 involves distal duplication with a normal metatarsal.
- Class 2 features a block metatarsal.
- Class 3 presents a Y-shaped metatarsal.
- Class 4 has a T-shaped metatarsal.
- Class 5 exhibits a normal metatarsal shaft with a widened head.
- Class 6 involves complete duplication, including the metatarsal.[12]
Evaluation
Polydactyly can be detected during a prenatal ultrasound, but the formal diagnosis is typically made at birth. Evaluation is performed in the neonatal period before any signs of dysfunction become apparent. Infants with polydactyly undergo a comprehensive physical examination and radiographic assessment to accurately classify their specific condition, which in turn aids in guiding surgical planning. Surgery aims to restore the proper shape, size, and functionality of the affected digit.[12][13] In the evaluation process, healthcare professionals must also assess whether the polydactyly is associated with a broader syndrome.
Polydactyly is often associated with a variety of syndromes, as mentioned below.
Bardet-Biedl Syndrome
Bardet-Biedl syndrome is an autosomal recessive disorder characterized by the gradual deterioration of night and peripheral vision due to retinitis pigmentosa. Individuals affected by this condition may additionally present with obesity, developmental disabilities, kidney disease, syndactyly, and polydactyly in the fingers and toes. The diagnosis is confirmed through genetic testing.
Greig Cephalopolysyndactyly Syndrome
Craniofacial abnormalities and supernumerary digits are distinctive features of Greig cephalopolysyndactyly. Individuals with this condition exhibit frontal bossing, ocular hypertelorism, an unusually broad nasal bridge, and an enlarged forehead. Moreover, the presence of extra digits, webbing, and fusion of digits are additional associated findings.
Short-Rib Polydactyly Syndrome
Short-rib polydactyly syndrome is a lethal autosomal recessive disorder distinguished by skeletal dysplasia, hypoplastic thorax, major organ anomalies, and pre- and postaxial polydactyly. Diagnosis can often be established through prenatal ultrasound.
Smith-Lemli-Opitz Syndrome
Autosomal recessive Smith-Lemli-Opitz syndrome is characterized by microcephaly, intellectual disabilities, behavior problems, syndactyly of the second and third toes, polydactyly, and malformations affecting the lungs, kidneys, genitalia, and gastrointestinal tract. Smith-Lemil-Opitz syndrome is caused by an abnormality in the DHCR7 (7-dehydrocholesterol reductase) gene. This mutation prevents cells from producing adequate amounts of cholesterol, leading to an accumulation of byproducts from cholesterol metabolism.
McKusick-Kaufman Syndrome
McKusick-Kaufman syndrome is characterized by postaxial polydactyly, congenital heart disease, hydrometrocolpos in females, and hypospadias and cryptorchidism in males. In instances of hydrometrocolpos, there is an accumulation of secretions within the vagina or extending up to the uterus, primarily caused by the accumulation of cervical secretions due to maternal estrogen stimulation. In female infants, there is a notable presentation of a large cystic abdominal mass emerging from the pelvis caused by dilatation of the vagina and uterus. Diagnosis can be initially established through clinical evaluation and subsequently confirmed through genetic testing.
Pallister-Hall Syndrome
Polydactyly, a bifid epiglottis, decreased pituitary function, imperforate anus, and a hypothalamic hamartoma are all characteristic features of Pallister-Hall Syndrome (PHS). PHS is an autosomal dominant disorder attributed to abnormalities in the GLI3 gene.
Fanconi Anemia
Fanconi anemia results from genetic mutations in genes responsible for DNA repair. Individuals with this condition are prone to developing aplastic anemia or acute myeloid leukemia and face an elevated risk of solid tumors. Common clinical anomalies associated with this condition include short stature, skin pigmentation abnormalities, and microphthalmia. Furthermore, affected individuals may exhibit various thumb abnormalities, such as absence, hypoplasia, bifid or duplication, rudimentary development, or a triphalangeal configuration.
Holt-Oram Syndrome
Holt-Oram syndrome, also known as heart-hand syndrome, is characterized by a combination of upper extremity and cardiac anomalies. Typically, the cardiac abnormality manifests as an atrial or ventricular septal defect. In addition to these heart-related issues, all individuals with this syndrome will present with fused carpal bones alongside various other phenotypes, including triphalangeal or absent thumbs.
Down Syndrome
Down syndrome or trisomy 21 is notably linked to the duplication of the first digit. This condition is also associated with a range of other characteristic features, including intellectual disabilities, congenital heart defects, hypothyroidism, early onset dementia, hearing loss, the presence of epicanthal folds, and increased spacing between the first and second toes.
Treatment / Management
The primary objective of treatment is to enhance hand function and cosmesis.[9] Therefore, surgical intervention may be pursued as necessary to achieve these objectives. The anticipated function of the supernumerary digit plays a crucial role in determining whether surgical intervention or nonoperative management is preferred. Nonoperative management may be viable when the digit possesses fully developed skeletal components and is not anticipated to disrupt hand function. Numerous surgical techniques are available for the management of hand polydactyly. Essential principles to be followed include the preservation of viable skin flaps, preservation of ligamentous structures to ensure finger stability, and meticulous surgical planning to uphold a robust pinch grip.[13]
Preaxial Polydactyly
Certain experts recommend surgical intervention for preaxial polydactyly between 7 and 12 months.[14] A general consensus exists to conduct surgery between 12 and 18 months to achieve the best functional outcomes. Operative interventions include digit ablation, the Bilhaut-Cloquet procedure, or an on-top plasty.[9] Surgical ablation is the most common choice as this procedure aims to conserve the more dominant thumb, ablate the smaller and more hypoplastic thumb, and reconstruct the collateral ligaments as necessary.[9] The objective is to establish a stable and functional thumb while minimizing the necessity for future repeat procedures. Simple suture ligation, involving tying a thread around the extra digit to cut off blood supply, is discouraged for managing preaxial polydactyly due to numerous potential complications, as discussed below.[13]
For patients with Wassel types I and II polydactyly, the Bilhaut-Cloquet procedure involves zig-zag incisions on the volar and dorsal surfaces of the bifid thumb. Subsequently, the central portions of the 2 thumbs are removed through osteotomy. The remaining osseous structures are secured with K wires, promoting the healing of both bone and soft tissues. This technique typically results in the formation of a thumb that is more normal in size and functionality. This procedure is recommended for patients with 2 symmetrical hypoplastic thumbs and involves merging these 2 digits into a single functional thumb.[15]
The Lamb, Marks, and Bayne technique is particularly valuable for addressing asymmetrical bifid thumbs. This procedure is especially advantageous for Wassel types III to VI polydactyly patients. In these cases, one of the thumbs typically takes on a dominant role, while the other, often the radial digit, exhibits hypoplasia and is subsequently excised. While excising the radial and ulnar digits, the surgeon carefully protects the abductor pollicis brevis and adductor pollicis. This is followed by removing the collateral ligament and periosteum from the base of the proximal phalanx. After the excision of the associated metacarpal bone, the remaining metacarpal is centralized to create a more functional digit. The collateral ligament and intrinsic muscles are attached to the proximal phalanx, and the metacarpophalangeal joint is stabilized using a K-wire. Following immobilization for a period of 4 to 6 weeks, the patient wears a protective splint for an additional 4 weeks to support the healing process.[16]
A typical thumb has 2 phalanges, whereas the triphalangeal thumb exhibits 3. A Wassel type I deformity involves an extra phalanx that takes the form of a wedge, leading to an angular deformity. This additional phalanx is called a delta phalanx, and the thumb remains opposable. On the other hand, a Wassel type II deformity results in a digit that is longer than usual and lies in the same plane as the other 4 fingers. A duplicated middle rectangular phalanx with atrophy of the thenar muscle renders the thumb nonopposable. Surgical goals for addressing Wassel type I deformity include correcting angulation, restoring length, enhancing web space development, and performing opponensplasty. In such cases, a Peimer reduction osteotomy can help preserve digit length. When dealing with a Wassel type II deformity, pollicization is typically performed as the surgical intervention.[17]
The on-top plasty is typically reserved for triphalangeal thumbs. In this procedure, the most functional distal portion of the thumb is transposed to the most suitable metacarpal among the available thumbs.[9] Similarly, in mirror hand/ulnar dimelia cases, the absent thumb is addressed through pollicization. The most dexterous radial finger is repurposed as the thumb, while the other supernumerary fingers are typically excised.[18]
Postaxial Polydactyly
Postaxial polydactyly is usually managed through surgical excision or suture ligation when no bony structures are present. However, surgical management with objectives akin to those in preaxial polydactyly becomes crucial if bony structures are involved. Establishing a structurally robust and functional digit is critical to achieving favorable outcomes. Therefore, usually, the less developed and more ulnar digit is excised. In cases of partial duplication, the transference of tendinous and ligamentous structures to the remaining digit plays a significant role in promoting good function.[9]
Central Polydactyly
Treatment for central polydactyly typically takes place around 12 months, and the primary objectives are to enhance aesthetics and function. Common surgical approaches often involve the excision or ligation of the more rudimentary digits. One guiding principle, supported by smaller studies of surgical outcomes, suggests prioritizing the creation of an opposable thumb and 3 functional digits over attempting to form a 5-fingered hand.[19][20]
Foot Polydactyly
Treating polydactyly of the foot similarly centers on optimizing function and cosmetic results. The fundamental principles for surgical intervention involve restoring a normal foot contour, preserving physeal alignment, and ensuring appropriate soft tissue tension around the toe joints.[12]
Differential Diagnosis
The diagnosis of polydactyly is quite evident in most cases, especially when the additional digit exhibits both bony and soft tissue features. However, when dealing with a more rudimentary digit that may only involve soft tissue, it is crucial to consider the possibility of infantile digital fibromatosis as part of the differential diagnosis.[21] Infantile digital fibromatosis is an uncommon, benign, recurring fibrous tumor located on the extensor aspects of the phalanges. This condition primarily affects the last 4 fingers or toes but typically spares the thumb and great toe. These lesions manifest as firm, reddish-pink, slow-growing nodules, typically reaching a size of about 2 cm. In most cases, the lesions will spontaneously resolve after approximately 12 months. However, treatment options are available if deemed necessary.
Prognosis
Polydactyly has aesthetic and functional implications. The primary concerns related to polydactyly revolve around its influence on fine motor development and overall functionality. With appropriate and well-timed intervention, most polydactyly cases result in favorable functional outcomes, leading to a positive overall prognosis.
Complications
Failure to treat polydactyly of the hand can result in reduced function and hindered fine motor skill development. In cases of preaxial polydactyly in the foot, a common complication is hallux varus, leading to joint pain and difficulty wearing footwear. Surgical treatment of polydactyly has various potential complications. There is a risk of developing a painful neuroma [13] or cyst formation in cases of suture ligation.[11] Treatment of polydactyly can be associated with several reported complications, including painful scarring, infection, joint instability, residual deformity, angulated growth, growth arrest, joint stiffness, and nail bed deformities.[11][13] One study reports a 19% revision rate for preaxial polydactyly, most commonly due to issues such as pain or instability.[22]
Postoperative and Rehabilitation Care
Patients who did not undergo bony osteotomy or ligament reconstruction can typically commence an active range of motion exercises within 10 to 14 days after the surgical procedure. Patients who undergo more extensive reconstruction can initiate a more gentle range of motion exercises with a hand therapist approximately 4 to 6 weeks after the surgery. Kirshner wires, if used, can typically be removed around 6 to 8 weeks following the operation. Gradually increasing usage is permitted approximately 6 to 8 weeks after the surgery while wearing a protective forearm-based thumb spica splint that includes the interphalangeal joint.
Deterrence and Patient Education
Polydactyly, also known as supernumerary digits, is a congenital anomaly of the hands or feet, characterized by an extra digit at birth. This condition can be inherited within families through an autosomal dominant pattern, which implies that only one parent carrying the abnormal gene is sufficient to pass on the anomaly to their offspring.
Timely evaluation and intervention are essential before any signs of dysfunction in the affected limb become evident. Swift action can lead to enhanced function and more favorable cosmetic outcomes. Although polydactyly often occurs as an isolated condition without any other associated anomalies, occasionally, it may be part of a syndrome.
X-rays are performed on patients with polydactyly to understand the anomaly's nature better. These X-rays are vital for categorizing the condition and planning of surgical procedures. Although surgical complications are infrequent, being aware of potential issues is crucial. These can encompass painful scarring, growth disruptions, and neuromas' development. Collaborating with the healthcare team is essential to provide the best care to patients and achieve optimal outcomes. Early intervention and a thorough evaluation are crucial in effectively addressing polydactyly and enhancing the overall quality of life for individuals affected by this condition.[11][13]
Enhancing Healthcare Team Outcomes
The clinical evaluation, management, and follow-up of individuals with supernumerary digits necessitate an interprofessional team approach. A primary care clinician typically makes a diagnosis early in life, and referral to an orthopedic specialist should occur promptly, if necessary, for early intervention. In cases requiring surgery, the team may involve professionals from anesthesiology, orthopedics, plastic surgery, as well as occupational and physical therapy. All medical professionals should be aware that polydactyly can be associated with a syndrome, and referral to various specialists may be necessary. Recognizing polydactyly is often associated with Fanconi anemia, which can lead to earlier diagnosis and referral, as well as improve morbidity and mortality rates.