Continuing Education Activity
Osteoblastoma is an uncommon benign bone-forming neoplasm that accounts for about 1% of all primary bone tumors. It commonly arises in the posterior elements of the spine and the sacrum. An accurate diagnosis of osteoblastoma is critical in determining the appropriate treatment modality and prognosis. In most cases, the basis for diagnosis is from clinical, radiological, and mainly histopathological examination. Osteoblastomas have a variable radiologic appearance ranging from indolent to very aggressive. This activity describes the cause, pathophysiology, presentation, and diagnosis of osteoblastoma and highlights the interprofessional team's role in its management.
Objectives:
- Review the etiology of osteoblastoma.
- Outline the evaluation process for a patient with suspected osteoblastoma.
- Explain the management options available for osteoblastoma.
- Summarize interprofessional team strategies for improving care coordination and communication to advance osteoblastoma treatment and improve outcomes.
Introduction
Osteoblastoma is an uncommon benign bone-forming neoplasm that accounts for about 1% of all primary bone tumors and 1 to 5 % of all benign bone tumors, and 10% of all osseous spinal neoplasms.[1][2][3] Historically it was referred to as giant osteoid osteoma highlighting its histopathologic similarities to osteoid osteoma.[4] In fact, some authors consider the entities to be variant expressions of the same pathologic process; however, the prevailing opinion is that they are distinct pathologic entities with varying clinical presentations. Osteoblastoma commonly arises in the posterior elements of the spine and the sacrum (approximately 30 to 40%).[5] Other common locations include the mandible (referred to as cementoblastoma) and long tubular bones (lower > upper extremities), where it is usually observed in the metadiaphysis.[4] An accurate diagnosis of osteoblastoma is critical in determining the appropriate treatment modality and prognosis.[6]
In most cases, the basis for diagnosis is from clinical, radiological, and mainly histopathological examination. Osteoblastoma has a variable radiologic appearance ranging from indolent to very aggressive.[7] In general, osteoblastoma has a good prognosis, and patients are often cancer-free after surgical treatments of intralesional curettage or marginal en bloc resection.[8] In cases that are not amenable to surgical excision, radiotherapy can be performed. In cases with more aggressive imaging and/or clinical features, distinction from aggressive (epitheliod) osteoblastoma or osteoblastoma-like osteosarcoma, two borderline osteoblastic tumors, is necessary.[9] In rare cases, osteoblastoma can be associated with systemic symptoms such as fever, weight loss, and diffuse periostitis, referred to as toxic osteosarcoma.[10]
Etiology
The exact etiology of osteoblastoma is still unknown, and so are the predisposing factors.
Epidemiology
Osteoblastoma is a rare benign neoplasm, accounting for 1% of all primary and 1 to 5 % of all benign bone tumors.[1] It affects mainly adolescents and young adults (mean age, 20 years) with a male predominance (2.5 to 1). The more aggressive forms are generally seen in slightly older patients, with a mean age of 33 years.[3][11] Osteoblastoma can involve any bone; however, it has a predilection for the spine and the sacrum (40 to 55%), most commonly involving the posterior elements of the spine.[12] Other common sites of involvement include the maxillofacial region's osseous structures and the long bones with a predilection for the lower extremity.[4]
Osteoblastoma less commonly involves the tarsal bones (talus and calcaneum), with case reports of involvement of the carpal bones and phalanges.[13][14] Like osteoid osteoma, osteoblastoma can be cortical, medullary, or rarely periosteal in a bone location.[15]
Pathophysiology
Like osteoid osteomas, osteoblastoma demonstrates marked new bone formation, although this process is generally more exuberant in osteoblastoma, which also tends to be more vascularized lesions.[16] The nidus within osteoblastoma tends toward less organized osteoid and trabecular bone, less abundant nerve fibers, and lacks prostaglandins shown in osteoid osteoma.[8][17] Although osteoblastoma is a benign tumor, it often causes significant bone destruction, infiltration of the soft tissues, and epidural extension. They usually show aggressive behavior with extensive uncontrollable local recurrence. Malignant transformation and metastatic diseases have been reported with osteoblastoma.[3][18]
The lack of prostaglandin production likely accounts for the variation in clinical presentation between osteoid osteoma and osteoblastoma, mainly the nighttime pain relieved by salicylates, which are pathognomonic for osteoid osteoma. In contrast, osteoblastoma is more often asymptomatic incidental lesions or presents as dull, localized pain, which rarely interferes with sleep.[19][20] Both osteoid osteoma and osteoblastoma express Runx2 and Osterix, transcription factors involved in osteoblastic differentiation.[21]
Histopathology
Macroscopic Examination (Gross Pathology)
Not surprisingly, osteoid osteoma and osteoblastoma have a similar macroscopic (gross) appearance in keeping with the radiologic presentation, which is also similar. Osteoblastoma has a copious vascular supply and appears red or red-brown, often with a gritty or sandpaper consistency. On curettage, the lesion may bleed profusely due to this rich vascularization. The average size is 3 to 3.5 cm, with a maximum diameter of up to 15 cm.[3][12]
In keeping with its radiographic appearance, the tumor is usually round to oval with a thinned, expanded cortex, surrounding sclerosis, and periosteal reaction. The border between the tumor and medullary cavity is sharp, often with some reactive bone. The tumor has a ''pushing'' border against the endosteal cortical surface and trabecular bone of the marrow, consistent with its benign nature versus an infiltrative or permeative margin seen with more aggressive or malignant lesions.[12] There may be evidence of hemorrhage, cystic changes with blood-filled spaces, and reparative changes in lesions with secondary aneurysmal bone cysts or large lesions.[22]
Histopathological Examination
The formation of osteoid and immature bone trabecula is a key feature of both osteoblastoma and osteoid osteoma; however, osteoblastoma generally produces more osteoid and is more vascularized with a less organized appearance of the nidus. A rare cartilaginous matrix may be present.[23] The tumor is composed of woven bone spicules or trabeculae. These spicules are haphazardly arranged and have a lining of a single layer of osteoblasts. The vascularity is rich, often with extravasated erythrocytes. While the mitotic rate may be high atypical mitoses are not present. Diffusely scattered osteoclast-type, multinucleated giant cells are often present.[12]
Osteoblastoma does not infiltrate or permeate preexisting lamellar bone structures as do osteosarcomas. Instead, they are separated by a distinct, narrow layer of bone-free fibrous tissue. Particular attention should be given to the border between the lesion and the preexisting cortex or marrow trabeculae to distinguish osteoblastoma from osteosarcoma. As described above, secondary aneurysmal bone cysts can occur, most commonly in large or expanded lesions.
Immunohistochemistry
Beta-catenin can be used to differentiate between osteoblastoma and osteosarcoma. Osteoblastoma is characterized by nuclear beta-catenin staining, whereas cytoplasmic or membranous staining of beta-catenin suggests osteosarcoma.[24]
A recent study has recommended using hypoxia-related microRNA-210 as a successful diagnostic marker for discriminating osteoblastoma and osteosarcoma.[25]
History and Physical
Osteoblastomas generally grow slowly with minimal or no symptomatology. In these patients, the lesions are incidentally found during imaging for the evaluation of other disease processes. This lack of symptomatology is in contradistinction from osteoid osteoma, which generally presents in slightly younger patients with night-time pain relieved by salicylates.[19][26]
When patients are symptomatic, they most commonly experience dull, localized pain. In some cases, the lesions are tender to palpation and present with associated soft tissue swelling, particularly when the lesion is close to the surface.[19] These mild, nonspecific symptoms may result in delayed presentation, with one study showing a median duration of symptoms of 6 months before presentation to a clinician.[19]
Osteoblastomas of the spine have similar symptoms to that of osteoid osteoma, namely back pain, scoliosis, and nerve root compression.[26][27] Nerve root compression may lead to muscle weakness or paraplegia.[28] Toxic osteoblastoma is a rare variant of osteoblastoma associated with systemic symptoms, including fever, anorexia, weight loss, and diffuse periostitis.[29]
Osteoblastomas rarely extend into the surrounding soft tissues and generally do not illicit surrounding soft tissue edema or inflammatory response. Osteoblastomas generally have a favorable prognosis, although local recurrence has been reported to be as high as 25%.[30] There have been rare case reports of malignant degeneration over the years, although recent genomic studies contradict these reports.[31][32]
Evaluation
Several imaging modalities are available for establishing the diagnosis of osteoblastoma. They include:
- Plain radiographs
- Computed tomography (CT)
- Magnetic resonance imaging (MRI)
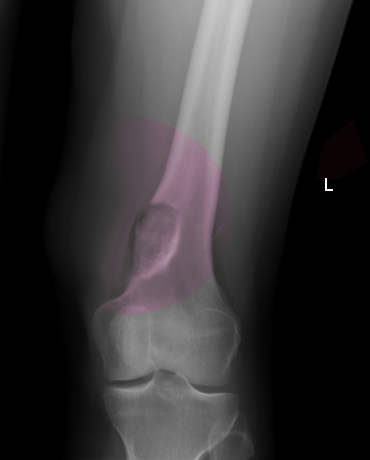
On imaging, osteoblastoma may be challenging to distinguish from osteoid osteoma.[7] The lesions are most often radiolucent, round to oval, with well-defined margins and reactive sclerosis.[3] It has been speculated that the imaging appearance may vary with patient age and lesion maturity, with lesions more often appearing radiolucent in young patients and increased sclerosis/ossification in older patients. The lesions often show an expanded appearance with thinning of the cortex; however, destruction or disruption of the cortex is only rarely identified (20% of cases).[3] In such cases, the lesions can be mistaken for a malignant process. Benign periosteal reaction is a common imaging feature present in up to 86% of cases.[3] In general, the lesions involving the spine, pelvis, and talus demonstrate less surrounding reactive bone formation compared to lesions of the long bones.[20]
Radiographs (X-ray)
Plain film radiography is generally the basis for the diagnosis of osteoblastoma, although the radiographic appearance can vary slightly, with four distinctive radiographic presentations having been described.[3][20]
- A similar radiographic appearance to osteoid osteoma but larger in size (>2 cm), with less reactive surrounding sclerosis and more overlying periostitis.
- Blown-out expansile lesion mimicking an aneurysmal bone cyst - this pattern is most commonly appreciated in lesions of the axial skeleton.[33]
- Aggressive appearing lesions mimicking a malignant process with cortical expansion, thinning, or disruption, as well as extensive periostitis and large size (often greater than 4 cm); aggressive osteoblastomas generally fall into this category; however, some authors believe aggressive osteoblastomas to be a distinct entity as opposed to a subtype of osteoblastoma.[9][34]
- Juxtacorticl (periosteal) lesions are exceedingly rare, comprising 8 of 62 osteoblastoma cases in the literature.[11][35] These lesions exhibit a thin periosteal margin but lack the common exuberant surrounding sclerosis seen in most lesions.[17]
Computed Tomography Scan
CT imaging serves a complementary role to plain-film radiography. The imaging appearance, not surprisingly, is similar to those described for radiography; however, CT can help further characterize the lesions, specifically the size, exact location, presence, or extent of cortical disruption and presence of a soft tissue component.[36] These findings are extremely helpful in treatment planning. CT is also helpful for characterizing lesions identified in locations that are suboptimally evaluated on radiographs due to overlapping/superimposed structures, such as lesions involving the spine or pelvis.[26]
Magnetic Resonance Imaging
MRI, like CT, can characterize the extent of the lesion and presence of aggressive imaging features, although CT better identifies the type of internal matrix (chondroid, fibrotic, or osteoid) of the lesions as well as the presence and extent of endosteal scalloping and cortical disruption. The exquisite contrast resolution on MRI allows for identifying reactive soft tissue edema and often better evaluates the soft tissue component if present. As previously described, perilesional edema (osseous or soft tissue) and extension into the surrounding soft tissues are rare with osteoblastoma. However, there have been case reports of lesions exhibiting these unusual imaging features leading to a misdiagnosis of more aggressive processes such as lymphoma or Ewing sarcoma.[37]
While not the primary modality for imaging evaluation of osteoblastoma, it is important to be aware of its imaging appearance on MRI, given that lesions of the spine can present with back pain, which is commonly evaluated with MRI. Generally, osteoblastomas have low to intermediate T1 signal intensity and intermediate to high T2 signal intensity. The areas of sclerosis will present as T1 and T2 hypointense regions. Areas of peritumoral edema will be T2 hyperintense and T1 hypointense. These imaging features are common findings in the majority of osseous neoplasms.[37][19]
Nuclear Medicine Imaging
Although not generally employed to evaluate osteoblastomas, studies using technetium-99m will demonstrate increased uptake in the mass corresponding to the osteoid formation within the lesion, and FDG-PET studies have revealed a high uptake in the tumor despite its pathologically benign features.[38]
Treatment / Management
The primary treatment of patients with osteoblastoma is surgery, either en bloc resection or curettage, depending on the clinical situation, location within the bone, and suspicion of malignancy.[39][40] En bloc resection is the preferred treatment when possible, resulting in a lower risk of local recurrence than curettage. Precise localization intraoperatively and aggressive complete resection can result in satisfactory clinical and radiological outcomes and is the key to preventing recurrence.[2] Recurrence rates have been reported as high as 25%. In some cases, multiple episodes of local recurrence may occur. There is no definite role for adjuvant chemotherapy or radiotherapy.[30][41]
Imaging surveillance may be necessary due to the risk of local recurrence. Although case reports exist in the literature on malignant degeneration, recent genomic studies contradict these findings.[29][30]
Differential Diagnosis
Histological Differential Diagnosis
- Osteoid osteoma
- Aneurysmal bone cyst
- Giant cell tumor of bone
- Osteoma with osteoblastoma-like features
- Osteoblastoma-like osteosarcoma[12]
Radiological Differential Diagnosis
- Osteoid osteoma
- Aneurysmal bone cyst
- Infection (Brodie abscess)
- Osteosarcoma
- Metastasis[7]
Osteoid Osteoma
As discussed above, osteoid osteoma and osteoblastoma can have a similar appearance both in imaging studies and histology. Osteoblastoma most commonly involves the spine and craniofacial structures (specifically the mandible), while osteoid osteoma is more common in the appendicular skeleton. While both can present with pain, osteoblastoma is most commonly asymptomatic or only mildly symptomatic, is not relieved by salicylates, or is disruptive to sleep, contrary to osteoid osteomas. Osteoid osteomas have been documented to resolve/regress spontaneously and, therefore, in some cases, are treated symptomatically, while others are treated with percutaneous radiofrequency ablation. In contrast, osteoblastoma has not been shown to resolve/regress; degeneration into osteosarcoma has been reported, although this has been disputed.[32][42]
One of the main differential characteristics is size, with osteoblastoma commonly measuring greater than 2 cm, while osteoid osteomas are smaller lesions. On pathology, there can be subtle differences in the nidus' appearance, which may allow for distinguishing osteoid osteoma from osteoblastoma. The nidus of osteoid osteoma tends to be more organized with a thin peripheral fibrovascular rim and a zonal pattern with central mineralization. Conversely, osteoblastoma generally lacks the fibrovascular rim and exhibits a lobulated or multifocal appearance.[8][43]
Aneurysmal Bone Cyst
Aneurysmal bone cysts (ABC) can occur as a primary benign osseous lesion (approximately 81%) or secondary, arising from preexisting lesions such as osteoblastoma, giant cell tumor, chondroblastoma, and fibrous dysplasia, to name a few. The typical radiographic appearance is an eccentric medullary-based, expansile, lucent lesion with a thinned cortex and well-defined, thinned, sclerotic margins. ABCs most commonly occur in the metaphysis of long bones (50 to 60%) but also occur in the spine and sacrum (20 to 30%), predominately in the posterior elements, similar to osteoblastoma. While most commonly medullary-based, the lesions can be cortically (12 to 18%) or periosteal (7 to 8%) in location.[22][44]
Cross-sectional imaging (CT and MRI) will often demonstrate fluid-fluid levels and internal septations, distinguishing features from osteoblastoma. On pathology, ABCs will demonstrate multiple blood-filled sinusoidal spaces with fibrous septations/walls, which may contain osteoid tissue, mature bone collections, or hemosiderin or reactive foam cells. There may also be interspersed richly vascularized solid components containing numerous giant cells.[45] Unlike osteoblastoma, which is commonly resected, ABCs are most commonly treated with curettage and bone grafting.
Giant Cell Tumor
Giant cell tumors are common benign osseous neoplasms (18 to 23% of benign osseous tumors) involving the metaphysis of long bones extending to the epiphysis. They are often closely approximated to the articular surface with well-defined margins (narrow zone of transition) without significant surrounding sclerosis. There is a female predilection (3 to 1) and a peak incidence between 20 to 30 years of age.[46] These features are in contradistinction to osteoblastoma, which demonstrates a male predominance, a peak incidence in the second decade, diaphyseal-metaphyseal location, and sclerotic margins with reactive periostitis. Periostitis may be seen in the setting of giant cell tumors if there is an associated pathologic fracture. Like osteoblastoma, secondary ABC components are a common feature (14%).[46] Microscopically giant cell tumors demonstrate giant cells evenly distributed throughout spindle cell stromal tissue, only rarely showing small amounts of osteoid formation.[47]
Infection (Brodie abscess)
Brodie abscess is a subacute or chronic form of osteomyelitis most commonly occurring in pediatric patients prior to closure of the growth plate; however, it can occur in patients of any age. The process presents as a radiolucent lesion, most commonly in the metaphysis of tubular bones (the tibia is the most common). There is a male predilection (2 to 1). Clinical presentation is often nonspecific with patients presenting with pain and/or swelling (median duration of 12 weeks). Most patients are afebrile, and less than half demonstrate elevated inflammatory markers. The key imaging features in skeletally immature patients is a serpentine tract extending to the closest physis, periostitis, and adjacent soft tissue swelling with well-defined sclerotic margins. The penumbra sign, a rim of T1 hyperintense signal lining the abscess with rapid enhancement, is a distinguishing imaging feature on MRI.
Osteoblastoma-like osteosarcoma and Aggressive Osteoblastoma
These lesions likely represent unique borderline entities distinct from osteoblastoma as opposed to subtypes of osteoblastoma, although classification is controversial and somewhat unclear in the literature.[9] While these borderline lesions are exceedingly rare, differentiation from osteoblastoma can profoundly impact patient treatment and outcomes. When lesions presumed to be osteoblastoma on imaging are encountered, it is important to assess for the presence of imaging features such as poorly defined margins, cortical disruption, or involvement of the adjacent soft tissues. In such cases, there should be an increased concern for a more aggressive process than conventional osteoblastoma, and an open biopsy instead of a percutaneous image-guided biopsy may be warranted to establish the diagnosis as the initial biopsy was reported as benign in 82% of osteoblastoma-like osteosarcoma (OBLOS).[9]
In one study, only 36% of OBLOS lesions demonstrated aggressive imaging features, thereby illustrating the difficulty in diagnosing these lesions.[48] Also, progressive symptoms or new symptoms following osteoblastoma treatment are worrisome for one of these more aggressive borderline tumors.
Osteosarcoma
There are multiple subtypes of osteosarcoma with varied imaging and clinical presentations, which is beyond the scope of this article. We will only briefly discuss a few subtypes (conventional and telangiectatic) and describe differentiating characteristics compared to osteoblastoma. In general, osteosarcoma will have a more aggressive appearance than osteoblastoma. The aggressiveness of a lesion is often first characterized by radiographic imaging. Radiographic features that must be evaluated to determine the aggressiveness of a lesion include margin, periostitis, endosteal scalloping, and cortical disruption. Non-aggressive lesions will generally have a well-defined "narrow zone of transition" with or without a sclerotic margin, while aggressive lesions will have incomplete or poorly defined margins "wide zone of transition." A classification scheme has been developed using the margins of lesions to help characterize the aggressiveness and risk of malignancy, and establish a differential diagnosis, termed the modified Lodewick-Madewell classification. Employing this scheme, lesions with well-defined sclerotic or non-sclerotic margins (IA and IB lesions) will be benign in 94% of cases; osteoblastoma most commonly falls into one of these categories, while osteosarcoma will most commonly be classified as type II or III lesions, those with incomplete or poorly defined margins, with a much higher risk of malignancy.[49]
Osteoblastoma will only demonstrate overlying periostitis in the setting of a pathologic fracture, in which case the patient will likely present with a history of recent injury. In contrast, osteosarcomas will present with aggressive forms of periostitis, such as an "onion-skin or sunburst" appearance. While osteoblastoma and osteosarcomas may both present with an expansile appearance with cortical thinning and endosteal scalloping, the cortex will be intact with osteoblastoma unless there is a pathologic fracture. Additionally, osteoblastoma will not have a soft tissue component, which is commonly present with osteosarcoma. Telangiectatic osteosarcoma, like conventional osteosarcoma, will have a more aggressive appearance than most osteoblastoma. Telangiectatic osteosarcoma will present as an expansile, lucent lesion with thin septations similar to ABCs.
It is important to remember that osteoblastoma can have secondary ABC components, mimicking ABC or telangiectatic osteosarcoma. On cross-sectional imaging (CT and MRI), fluid-fluid levels will be present, also similar to ABCs. As previously stated, osteoblastoma should not have aggressive periostitis, cortical disruption, or enhancing internal soft tissue components, all features of telangiectatic osteosarcoma. In cases where the lesion's aggressiveness is in question, as may be the case with the borderline lesions described above, there should be a high clinical suspicion for osteosarcoma. Open biopsy, repeat biopsy due to sampling error, or expert consultation may be considered in such cases to ensure the appropriate diagnosis is obtained and the patient receives the appropriate medical/surgical treatment. On histopathology, osteosarcomas should demonstrate highly pleomorphic cells producing osteoid with mitotic figures and areas of necrosis with a permeative pattern invading surrounding bone. In complex cases where the diagnosis remains in question, immunohistochemistry may help differentiate osteosarcoma from osteoblastoma or osteoid osteoma. Osteoid osteoma and osteoblastoma tumor cells will be positive for the FOS marker, which will be negative in osteosarcoma.[5][50][51]
Prognosis
The prognosis of osteoblastoma is excellent, with most patients cured following the initial surgical treatment. However, local recurrence is a relatively common complication, with rates ranging from 15% to 25%.[12]
Recurrence is more common in the setting of lesions treated with curettage. Since spinal lesions are often treated with curettage due to the anatomic challenges and morbidity of performing wide local excision, it is not surprising that high recurrence rates have been associated with spinal lesions. Recurrence is most common in the initial two years following treatment and rare beyond two years post-treatment. Also, borderline lesions such as aggressive osteoblastoma or osteoblastoma-like osteosarcoma can be mistakenly diagnosed as osteoblastoma, leading to delayed diagnosis and incomplete treatment. Long-term follow-up imaging and clinical surveillance (generally at least two years based on recurrence data) are commonly performed due to the risk of recurrence.[41]
While degeneration into osteosarcoma has been reported in the literature, recent genomic studies contradict these findings, and malignant transformation remains controversial. Degeneration of osteoblastoma to osteosarcoma is exceptional but has been reported[3][12]
Complications
The most frequently encountered immediate postoperative complications following osteoblastoma surgical removal are:
-
Wound infections, as well as infections involving the urinary tract and lungs
-
Surgical site hemorrhage
-
A loss of stability involving the surgical stabilization construct
Tumor recurrence typically occurs late (i.e., months to years) after the procedure, and the clinician and patient must plan for this possibility for a scheduled period following the procedure. No universally accepted time intervals exist to define this period during which follow-up should occur; therefore, the range varies significantly. It is probably beneficial to continue these studies until the likelihood of regrowth of this tumor, as reported in the literature, is almost negligible.
Deterrence and Patient Education
Osteoblastoma recurs in approximately 10 to 20% of patients; the odds of tumor recurrence correlate with how well the surgeon completely removed the tumor without damaging normal structures. The time needed before the patient can return to normal daily activities will vary based on the tumor's location and the procedure used to remove it. Should the tumor recur, treatment still utilizes the same methods.
Enhancing Healthcare Team Outcomes
Osteoblastoma is ideally managed by an interprofessional team that consists of orthopedists, radiologists, and pathologists. Multidisciplinary conferences which allow for correlation between gross, radiographic, and microscopic features of the lesion are crucial to establishing the definitive diagnosis of osteoblastoma, ensuring timely and appropriate treatment. Postoperatively, patients may require physical therapy depending on the location of the lesion and the extent of surgical treatment, as well as long-term follow-up due to the possibility of tumor recurrence. Initially, follow-up is performed by the orthopedic oncology team; however, since recurrence is rare beyond two years, patients can be followed by primary care after two years with an orthopedic referral for any new symptoms. This interpr4ofessional approach will result in improved patient outcomes with appropriate vigilance for recurrence. [Level 5]