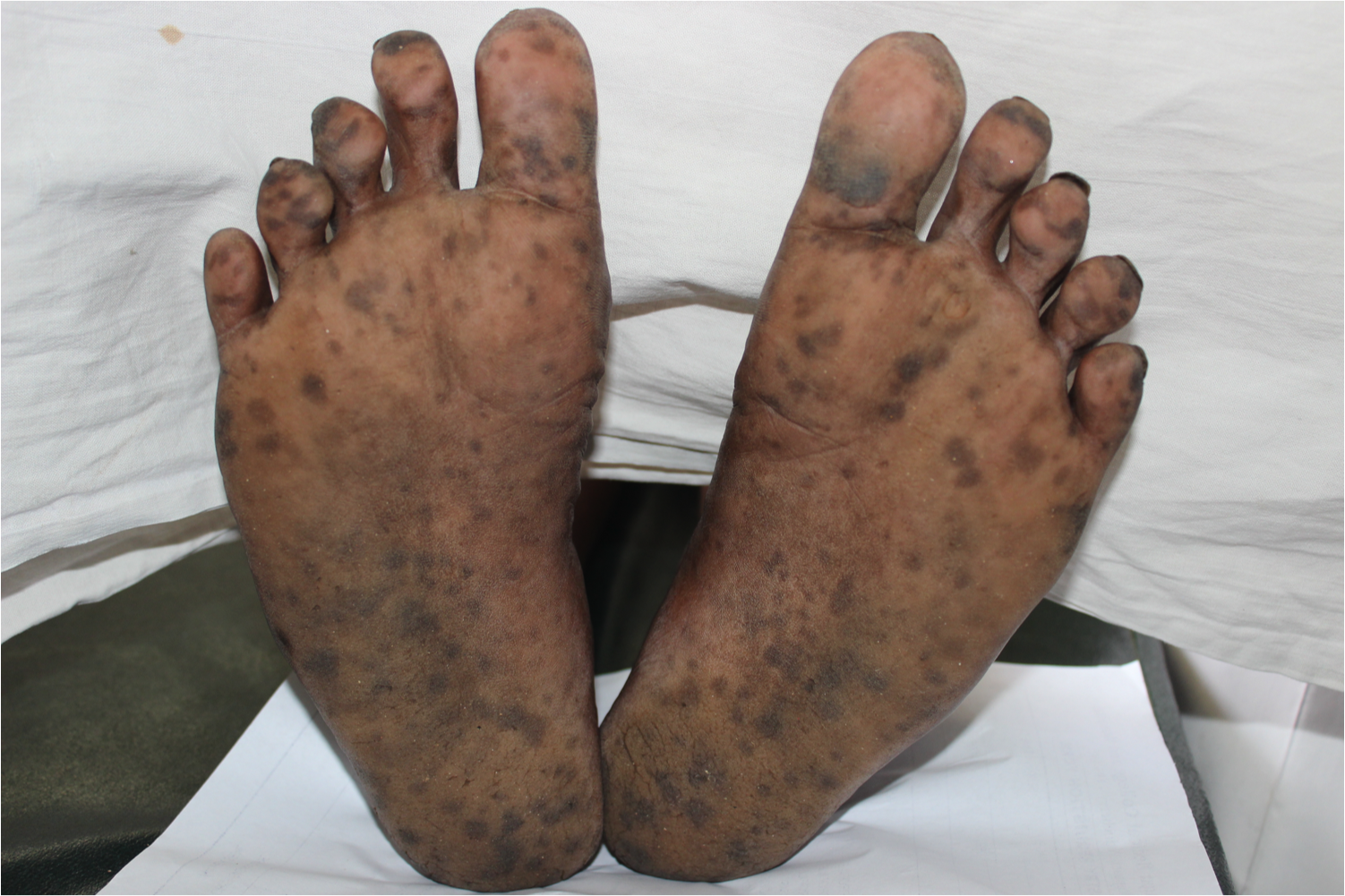
[1]
Wei Z, Li GY, Ruan HH, Zhang L, Wang WM, Wang X. Laugier-Hunziker syndrome: A case report. Journal of stomatology, oral and maxillofacial surgery. 2018 Apr:119(2):158-160. doi: 10.1016/j.jormas.2017.12.003. Epub 2017 Dec 12
[PubMed PMID: 29246753]
Level 3 (low-level) evidence
[3]
Montebugnoli L, Grelli I, Cervellati F, Misciali C, Raone B. Laugier-hunziker syndrome: an uncommon cause of oral pigmentation and a review of the literature. International journal of dentistry. 2010:2010():525404. doi: 10.1155/2010/525404. Epub 2010 Jul 7
[PubMed PMID: 20671949]
[4]
Moore RT,Chae KA,Rhodes AR, Laugier and Hunziker pigmentation: a lentiginous proliferation of melanocytes. Journal of the American Academy of Dermatology. 2004 May
[PubMed PMID: 15097932]
[5]
Nayak RS, Kotrashetti VS, Hosmani JV. Laugier-Hunziker syndrome. Journal of oral and maxillofacial pathology : JOMFP. 2012 May:16(2):245-50. doi: 10.4103/0973-029X.99079. Epub
[PubMed PMID: 22923898]
[6]
Sachdeva S, Sachdeva S, Kapoor P. Laugier-hunziker syndrome: a rare cause of oral and acral pigmentation. Journal of cutaneous and aesthetic surgery. 2011 Jan:4(1):58-60. doi: 10.4103/0974-2077.79199. Epub
[PubMed PMID: 21572687]
[7]
Makhoul EN, Ayoub NM, Helou JF, Abadjian GA. Familial Laugier-Hunziker syndrome. Journal of the American Academy of Dermatology. 2003 Aug:49(2 Suppl Case Reports):S143-5
[PubMed PMID: 12894104]
[8]
Duan N,Zhang YH,Wang WM,Wang X, Mystery behind labial and oral melanotic macules: Clinical, dermoscopic and pathological aspects of Laugier-Hunziker syndrome. World journal of clinical cases. 2018 Sep 26
[PubMed PMID: 30283795]
Level 3 (low-level) evidence
[9]
Lampe AK, Hampton PJ, Woodford-Richens K, Tomlinson I, Lawrence CM, Douglas FS. Laugier-Hunziker syndrome: an important differential diagnosis for Peutz-Jeghers syndrome. Journal of medical genetics. 2003 Jun:40(6):e77
[PubMed PMID: 12807976]
[10]
Wang WM, Wang X, Duan N, Jiang HL, Huang XF. Laugier-Hunziker syndrome: a report of three cases and literature review. International journal of oral science. 2012 Dec:4(4):226-30. doi: 10.1038/ijos.2012.60. Epub 2012 Nov 23
[PubMed PMID: 23174847]
Level 3 (low-level) evidence
[11]
Asati DP, Tiwari S. Laugier-Hunziker syndrome. Indian journal of dermatology, venereology and leprology. 2011 Jul-Aug:77(4):536. doi: 10.4103/0378-6323.82422. Epub
[PubMed PMID: 21727718]
[12]
Jabbari A,Gonzalez ME,Franks AG Jr,Sanchez M, Laugier Hunziker syndrome. Dermatology online journal. 2010 Nov 15
[PubMed PMID: 21163174]
[13]
Wondratsch H, Feldmann R, Steiner A, Breier F. Laugier-hunziker syndrome in a patient with pancreatic cancer. Case reports in dermatology. 2012 May:4(2):174-6. doi: 10.1159/000342070. Epub 2012 Aug 16
[PubMed PMID: 22949900]
Level 3 (low-level) evidence
[14]
Ergun S, Saruhanoğlu A, Migliari DA, Maden I, Tanyeri H. Refractory Pigmentation Associated with Laugier-Hunziker Syndrome following Er:YAG Laser Treatment. Case reports in dentistry. 2013:2013():561040. doi: 10.1155/2013/561040. Epub 2013 Dec 3
[PubMed PMID: 24367727]
Level 3 (low-level) evidence
[15]
Abduljabbar T, Vohra F, Akram Z, Ghani SMA, Al-Hamoudi N, Javed F. Efficacy of surgical laser therapy in the management of oral pigmented lesions: A systematic review. Journal of photochemistry and photobiology. B, Biology. 2017 Aug:173():353-359. doi: 10.1016/j.jphotobiol.2017.06.016. Epub 2017 Jun 15
[PubMed PMID: 28641206]
Level 1 (high-level) evidence
[16]
Pereira PM,Rodrigues CA,Lima LL,Reyes SA,Mariano AV, Do you know this syndrome? Anais brasileiros de dermatologia. 2010 Sep-Oct
[PubMed PMID: 21152810]
[17]
Miličević T, Žaja I, Tešanović D, Radman M. Laugier-Hunziker syndrome in endocrine clinical practice. Endocrinology, diabetes & metabolism case reports. 2018:2018():. pii: EDM180025. doi: 10.1530/EDM-18-0025. Epub 2018 Jul 26
[PubMed PMID: 30087778]
[18]
Niiyama S, Katsuoka K. Laugier-Hunziker syndrome. European journal of dermatology : EJD. 2013 Apr 1:23(2):284-5. doi: 10.1684/ejd.2013.2000. Epub
[PubMed PMID: 23567753]
[19]
Barman PD, Das A, Mondal AK, Kumar P. Laugier-Hunziker Syndrome Revisited. Indian journal of dermatology. 2016 May-Jun:61(3):338-9. doi: 10.4103/0019-5154.182429. Epub
[PubMed PMID: 27293265]