Continuing Education Activity
Klippel-Feil syndrome is a congenital condition characterized by the abnormal fusion of 2 or more cervical vertebrae, which results in a shortened neck. This skeletal anomaly also manifests with facial asymmetry, low hairline, and limited neck mobility, leading to chronic headaches, restricted neck mobility, and neck muscle pain. Moreover, Klippel-Feil syndrome may predispose individuals to spinal stenosis, neurological deficits, and cervical spinal deformities and instability, with some cases presenting with multiple syndromes. Diagnosis typically involves a comprehensive evaluation, including physical examination, imaging studies, and, in some cases, genetic testing. Management strategies aim to address symptoms and complications by using conservative approaches such as physical therapy to enhance mobility and muscle strength and pain management with medications or injections.
Surgical interventions, such as decompression or stabilization procedures, aim to alleviate spinal cord and nerve pressure and improve spinal alignment in patients experiencing progressive symptoms. Long-term monitoring and interprofessional care are essential to effectively managing the condition's complications. This activity provides a comprehensive understanding of the condition's pathophysiology, presentations, and evidence-based diagnostic and treatment strategies. This activity also aims to enhance clinicians' proficiency in evaluating and managing Klippel-Feil syndrome by effectively collaborating within an interprofessional healthcare team dedicated to caring for patients with this condition.
Objectives:
Identify Klippel-Feil syndrome through comprehensive evaluation, including physical examination and imaging studies.
Implement evidence-based management strategies for Klippel-Feil syndrome, including conservative approaches and surgical interventions, to effectively address symptoms and complications.
Apply interdisciplinary care principles in the management of Klippel-Feil syndrome, collaborating with specialists from various medical fields to optimize patient outcomes.
Collaborate with allied multidisciplinary healthcare professionals, including physical therapists and orthotists, to provide comprehensive care for patients with Klippel-Feil syndrome.
Introduction
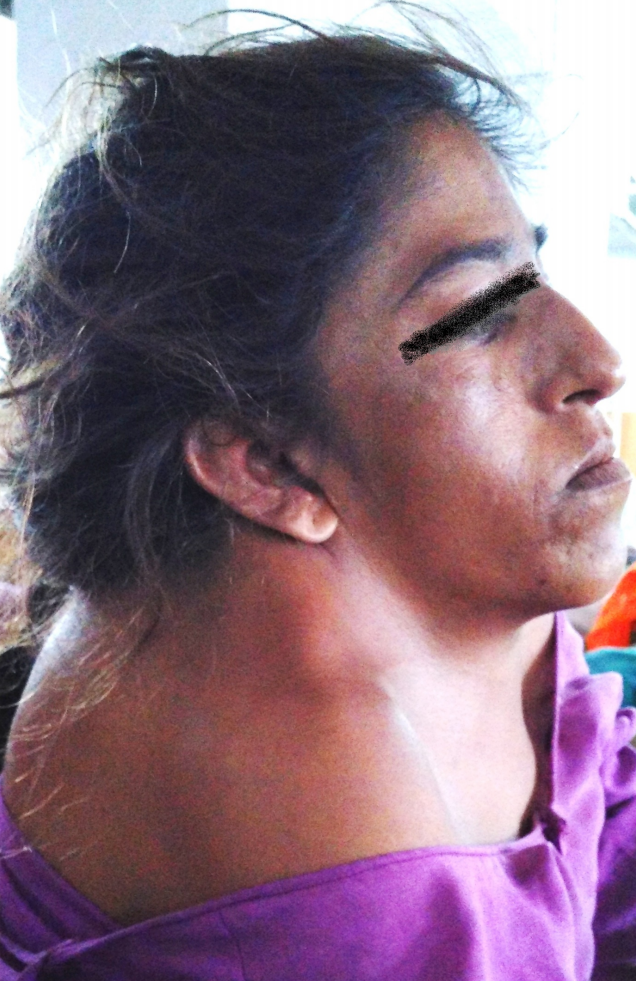
Klippel-Feil syndrome is a complex, congenital condition characterized by the abnormal fusion of 2 or more cervical vertebrae due to the failure of proper segmentation during early fetal development. This fusion results in a shortened neck due to the failure of proper segmentation during early fetal development, leading to congenital cervical vertebral fusion. The condition was first described by Maurice Klippel and Andre Feil in 1912.[1] Klippel-Feil syndrome presents with the classic triad of a short neck, low posterior hairline, and limited neck mobility (see Image. Classic Triad of Klippel-Feil Syndrome).
Since its initial description, Klippel-Feil syndrome has been associated with multiple spinal and extraspinal abnormalities.[2][3] These anomalies may contribute to chronic headaches, restricted neck motion, and neck muscle pain. Furthermore, Klippel-Feil syndrome can lead to spinal stenosis, neurological deficits, cervical spinal deformities, and instability. Additionally, the condition may present with various other congenital anomalies.[4][5]
Etiology
The etiology of Klippel-Feil syndrome is not well known.[6] Several studies have hypothesized that vascular disruption, global fetal insult, primary neural tube complications, or related genetic factors may carry implications in this condition's development.[7][8][9] Klippel-Feil syndrome may be present with fetal alcohol syndrome, Goldenhar syndrome, and Sprengel deformity.[10][11][12] Klippel-Feil syndrome can be caused by heritable mutations in the GDF6, GDF3, and MEOX1 genes. GDF6 and GDF3 influence embryonic bone development. The MEOX1 gene encodes the homeobox protein MOX1, which regulates vertebral separation. GDF6 and GDF3 abnormalities are inherited in an autosomal dominant pattern, while MEOX1 mutations are autosomal recessive.[13]
Epidemiology
Klippel-Feil syndrome affects approximately 1 in 40,000 to 42,000 newborns worldwide, with a slight female predominance. A study by Nouri et al demonstrated a 2.0% incidence of Klippel-Feil syndrome on magnetic resonance imaging (MRI) in a global cohort of 458 patients.[14][15] In addition, Brown et al reviewed 1400 skeletons and reported a 0.71% incidence of the condition.[16] Notably, Klippel-Feil syndrome may also be asymptomatic. Children who do not undergo cervical imaging or present with an obvious physical deformity are likely to grow into adulthood unaware of their condition.[17][18]
Pathophysiology
Faulty cervical spine segmentation occurs during weeks 3 to 8 of embryonic development, resulting in persistent fusion of the involved vertebrae.[19] The Samartzis classification scheme is frequently used to describe Klippel-Feil syndrome fusions.[20] Type I involves a single congenitally fused segment, whereas type II involves multiple, noncontiguous, congenitally fused segments. Type III consists of multiple, contiguous, congenitally fused segments. About 25% of patients with Klippel-Feil syndrome present with type I deformities, while 50% have type II, and another 25% develop type III deformities.
History and Physical
History
Klippel-Feil syndrome presents with varying manifestations, depending on the severity and location of cervical vertebral involvement. The classic Klippel-Feil syndrome triad is observed in less than 50% of patients. This variability may arise from challenges in accurately assessing congenitally fused cervical patterns over time and potential biases affecting the evaluation of the classic clinical Klippel-Feil syndrome triad. Recent research also indicates that many sporadic Klippel-Feil syndrome cases are discovered incidentally through radiographic evaluation, often revealing asymptomatic single, fused cervical segments.
Children with Klippel-Feil syndrome may be predisposed to congenital spinal stenosis. Consequently, these patients may sustain significant neurological deficits after experiencing a relatively low-impact or low-energy injury. Skeletal abnormalities can vary from minor deformities to distortions that affect both mechanical and neurological function. In severe cases, patients may present with torticollis, intubation difficulties, and pain insensitivity, leading to significant environmental injuries, such as burns.[21][22]
Visceral involvement and audiovisual impairment are additional features reported in Klippel-Feil syndrome cases. Cardiovascular anomalies are also noted, affecting 4.4% to 14% of individuals with the syndrome. The most common anomalies include ventricular septal defects, aortic coarctation, aortic arch hypoplasia, aortic root aneurysm, and abnormal pulmonary vessel insertion.[23]
Prenatal ultrasound or MRI examinations may reveal cervical spine anomalies in affected individuals.[24] In addition, birth history may indicate difficulties during delivery and neonatal care. Assessing the developmental history of children is crucial, as delays in reaching developmental milestones may be observed. Caregivers of patients may also report the presence of similar skeletal conditions in other family members.
Physical Examination
Notably, it is essential to thoroughly assess all organ systems during neonatal physical examinations as Klippel-Feil syndrome may be accompanied by various associated anomalies. A craniofacial examination may reveal abnormalities such as facial dysmorphism, microcephaly, or a low posterior hairline. Examination of the head and neck may reveal a short neck, limited neck mobility, and torticollis. Auscultation may detect murmurs if a cardiac disorder is present. Neurological signs can vary widely, with some patients showing no abnormalities while others may exhibit abnormal sensorimotor function and reflexes.
Neonates exhibiting skeletal abnormalities during physical examination should undergo further evaluation for associated anomalies. For instance, some newborns diagnosed with Klippel-Feil syndrome may require an auditory brainstem response test to assess for sensorineural deafness.[25][26] Abdominal sonography may be used to investigate potential visceral anomalies, such as renal ectopia and Mullerian duplication in infants.[27][28]
Older children and adults should undergo a comprehensive physical examination, particularly if symptom progression needs to be documented or if Klippel-Feil syndrome directly influences a new illness, such as a respiratory infection. It is essential for all patients suspected of having Klippel-Feil syndrome to undergo a thorough neurological examination to exclude sudden sensorimotor impairment and bowel or bladder incontinence.
Evaluation
Klippel-Feil syndrome often coexists with other congenital conditions such as Sprengel deformity, Duane syndrome, renal agenesis, Wildervanck syndrome, and various cardiovascular abnormalities. Approximately 50% of patients diagnosed with Klippel-Feil syndrome also exhibit concurrent scoliosis. In addition, around 50% of patients present with atlantoaxial instability, while approximately 30% experience renal disease and another 30% suffer from deafness. Laboratory and imaging tests are instrumental in identifying organ dysfunction in individuals with Klippel-Feil syndrome.
Ultrasonography
Ultrasonography allows noninvasive imaging of the intracranial cavity, cervical spine, and visceral organs in neonates. Besides detecting cervical vertebral fusion, ultrasound may reveal Klippel-Feil syndrome-related cardiac, gastrointestinal, and renal anomalies. Certain renal and genitourinary malformations, such as renal ectopia and uterine duplication, may not be evident on physical examination but may be found on ultrasonography.
Plain Radiography
Plain x-rays may be obtained in older patients with Klippel-Feil syndrome to assess the extent of cervical vertebral fusion and the involvement of the ribs and other spinal segments. The examination should include anteroposterior, lateral, and odontoid views in flexion and extension. These studies help to evaluate the stability of the atlantoaxial, atlantooccipital, and subaxial joints. The cervical spine must be thoroughly evaluated before procedures such as intubation, laryngoscopy, head manipulation, or intraoperative positioning due to the risk of atlantoaxial subluxation and craniovertebral dislocation.
Imaging the thoracic and lumbar spine is essential to assess for conditions such as scoliosis, spinal bifida, or hemivertebrae in individuals with Klippel-Feil syndrome. Additionally, the presence of a "wasp-waist" sign, indicating anteroposterior narrowing, may be observed. Flexion-extension x-rays can provide valuable insights into spinal stability and movement, particularly in clinically stable patients.
Computed Tomography
Cervical computed tomography (CT) offers a comprehensive assessment of spinal anatomy and bony structures, particularly bony fusion in Klippel-Feil syndrome. CT helps evaluate central canal stenosis through axial views, providing detailed insights into spinal pathology.
Magnetic Resonance Imaging
MRI is a valuable tool for evaluating the integrity of the spinal cord, disc space, nerve rootlets, ligaments, and other soft tissue structures in Klippel-Feil syndrome. This study can also detect associated spinal cord abnormalities, such as Chiari malformations and diastematomyelia. MRI is most useful in patients who present with neurological deficits.[29][30][31]
Laboratory Testing
Clinical findings should guide laboratory testing. For example, cardiovascular or renal dysfunction may render abnormal blood counts, metabolic panels, or arterial blood gases.[32] Tests such as C-reactive protein and erythrocyte sedimentation rate may help rule out inflammation as the cause of joint or bone pain.[33] Additionally, assessing the coagulation profile can be crucial to rule out clotting abnormalities before spinal corrective procedures.
Treatment / Management
Most patients with Klippel-Feil syndrome receive nonoperative management, except in cases of acute neurological deficits, cervical instability, or chronic neurological issues that pose risks, necessitating operative management.[34][35] The general treatment approaches to this condition are explained below.
Nonoperative Management
Treatment for Klippel-Feil syndrome typically involves conservative measures tailored to manage symptoms. Patients with 1- or 2-level fusions below C3 may undergo monitoring and nonoperative management. Patients can engage in contact sports such as hockey and rugby with appropriate education. However, individuals at higher risk of spinal deformity, particularly those with cervical fusion above C3, especially extending to the occiput, and long cervical spine fusions, should consider activity modification. Avoiding contact sports is crucial for these patients due to their increased risk of symptoms and susceptibility to spinal injuries.
Clinicians should remember that many individuals with Klippel-Feil syndrome have polysyndromic presentations. Pediatricians are critical in facilitating communication and coordination among various specialists to address cardiac, renal, or gastrointestinal congenital abnormalities, particularly in younger patients. Interprofessional collaboration among healthcare providers becomes even more vital, especially when patients are surgical candidates.
Operative Management
Patients with persistent neurological pain, myelopathy, new-onset muscle group weakness, and documented spinal instability are operative candidates.[36] Surgical decision-making is influenced by spinal deformities and instability. Depending on clinical evaluation, surgeons may opt for cervical fusion using either an anterior or posterior approach.
The anterior approach involves procedures such as anterior cervical fusion or corpectomy with synthetic or bone graft placement.[37] Cervical total disc arthroplasty is being investigated as a surgical option, showing promise in enhancing quality of life and preventing adjacent-level disease in adults with degenerative conditions. Posterior approaches, such as decompression and fusion, are viable options for treatment. In severe deformities, a combined anterior-posterior approach may be considered. Additionally, surgical or bracing intervention may be necessary for associated compensatory thoracic scoliosis.
Patients with hearing impairment may require otolaryngological evaluation. These assessments help determine the need for interventions such as cochlear implants or hearing devices.[38]
Differential Diagnosis
Various congenital conditions may present similarly to neonatal Klippel-Feil syndrome, often affecting multiple organ systems. These disorders include Mayer-Rokitansky-Küster-Hauser syndrome, vertebral defect, anal atresia, cardiac defect, tracheoesophageal fistula/esophageal atresia, renal and limb defects (VACTERL), and Wildervanck syndrome. Differentiating each condition is essential to short- and long-term management planning.
The differential diagnosis of Klippel-Feil syndrome in older children and adults should include healing osteomyelitis or discitis, prior fusion without instrumentation, juvenile idiopathic arthritis, juvenile rheumatoid arthritis, and ankylosing spondylitis. These conditions often manifest with symptoms such as cervical vertebral pain, immobility, or shortening, which may resemble atypical Klippel-Feil syndrome. A comprehensive clinical and diagnostic assessment is essential to differentiate these conditions from Klippel-Feil syndrome.
Prognosis
Patients with Klippel-Feil syndrome and cervical fusion above C3 tend to be more symptomatic. Research by Samartzis et al indicates that approximately two-thirds of individuals with this condition remain asymptomatic for over 8 years. Among those affected, individuals with type I deformity tend to experience more axial symptoms, while those with types II and III may develop myelopathy and radiculopathy.
Complications
Congenital cervical fusion may predispose individuals to various conditions, necessitating monitoring and prompt management. These conditions include fractures, adjacent segment disease, disc degeneration, spondylosis, spinal canal stenosis, disc herniation, and osteophyte formation. Timely intervention, including surgical management when necessary, can mitigate complications and improve outcomes for individuals with Klippel-Feil syndrome.
Deterrence and Patient Education
Patients and their relatives must receive genetic counseling to guide family planning. In addition, patients and their parents should be informed about support groups such as the Genetic and Rare Diseases Information Center and Klippel-Feil Syndrome Freedom. These resources offer education, management techniques, and support to help individuals optimize their life options based on the severity of the condition.
Standardized guidelines for sports participation are currently lacking. However, the proposed recommendations include the following:
- Sports participation is absolutely contraindicated in all patients with type I lesions or type II fusions with specific criteria, including limited motion range, C2 involvement, instability or spondylosis, and occipitocervical anomalies.
- Sports participation is relatively contraindicated in patients with type II lesions and a history of transient quadriplegia.
- Patients with type II lesions below C3, exhibiting adequate cervical spine motion and without instability or spondylosis, may engage in sports with appropriate education and precautions.[39]
Nonsurgical treatment approaches require an explanation to the patient and parents. Surgeons must establish realistic expectations regarding the procedure's achievable outcomes for surgical candidates.
Pearls and Other Issues
Klippel-Feil syndrome presents a complex array of diagnostic and management challenges. Although the Klippel-Feil syndrome triad includes a short neck, low posterior hairline, and limited neck mobility, more than half of patients do not exhibit this triad. Some individuals remain asymptomatic until adulthood, while others present polysyndromically. A thorough clinical evaluation with appropriate diagnostic testing can help guide proper treatment.
Most patients with Klippel-Feil syndrome are treated nonoperatively, except in cases of acute neurological deficit, cervical instability, or chronic neurological issues, which require operative intervention. Careful consideration is needed when advising patients with high cervical fusions due to the heightened risk of neurological injury. Early intervention with modification of physical activities and therapy may lower the risk of degenerative disc changes and trauma. While standardized guidelines for sports participation are lacking, recommendations offer insight into contraindications and relative risks associated with different lesion types, emphasizing the importance of individualized care in navigating the complexities of Klippel-Feil syndrome management.
Enhancing Healthcare Team Outcomes
Diagnosing and treating patients with Klippel-Feil syndrome requires an interprofessional approach involving a neurologist, orthopedic surgeon or neurosurgeon, pediatrician, nurse practitioner, and physical therapist, among other medical staff personnel. Orthotists are critical in crafting assistive devices for patients with functional limitations.
Depending on the presentation, the disorder may be managed by clinicians either nonsurgically or surgically. While several types of surgical procedures are available, their outcomes can be unpredictable, and the risk of severe complications is ever-present. Therefore, an interprofessional healthcare team of surgical specialists, including neurosurgeons, otolaryngologists, orthopedists, and oromaxillofacial surgeons, is essential for comprehensive care.[40][41][42] Interprofessional collaboration is the optimal approach to optimize outcomes for patients with Klippel-Feil syndrome.