Pathophysiology
Thick ascending limb of Henle
[DIAGRAM 1]
Bartter syndrome, which presents as a salt-wasting disease with hypokalemia, was initially described in 1962 and thought to be already a genetic disease.[1] Multiple cases were then described as such, and nephrologists started to study the genetics of the disease.
Early studies of renal physiology demonstrated that most of the lumen of the nephron has a negative charge except in the thick ascending limb of Henle, which led to the discovery of sodium and chloride transport in this part of the nephron. It appears that chloride first binds to the transporter, followed then by sodium and potassium. It is this first move of chloride that leaves a positive charge in the lumen. Researchers later identified this transporter as NKCC2.[2] Renal physiologists already knew at the time (Kokko, Imai) that loop diuretics (furosemide) inhibited this transporter. It appeared that the mutation of NKCC2 could not account for all the cases of Bartter syndrome. Hence, more studies followed.
Potassium is not abundant in this segment of the nephron and is however necessary for the transport of sodium and chloride by NKCC2. It felt intuitively that potassium needed to be recycled by the thick ascending limb cells into the lumen. Also, creating a more positive charge. That intuition led to the discovery of the ROMK channel.[3]
This potassium channel gets expressed at the luminal side of the cell and, when available, allow potassium to travel down their concentration gradient into the lumen. Any genetic defect of this channel will cause Bartter syndrome (now called Barter syndrome Type II).
That still did not account for all the patients with Bartter syndrome. The ingenuity of nephrology physiologists led to think that maybe chloride played a role in some cases of Bartter syndrome. Sure enough, researchers discovered a mechanism that transports chloride to the interstitium.[4] The responsible molecule is a chloride channel that is expressed at the apical side of the thick ascending cell and allows chloride to flow from the intracellular component to the interstitium of the renal cortex. A genetic mutation of that channel was described and caused Bartter Syndrome Type III. If most of the patients with Bartter syndrome fall in these three categories, there are still cases that cannot be explained by a mutation of one of these three genes. One example is an association found between Bartter syndrome and activation of the calcium-sensing receptor (CASR).[5] It appears that the constant activation of the CASR inhibits the function of the potassium channel (ROMK), as in Bartter Type II. It is most probable that more similar discoveries will come to account for all Bartter syndrome cases.
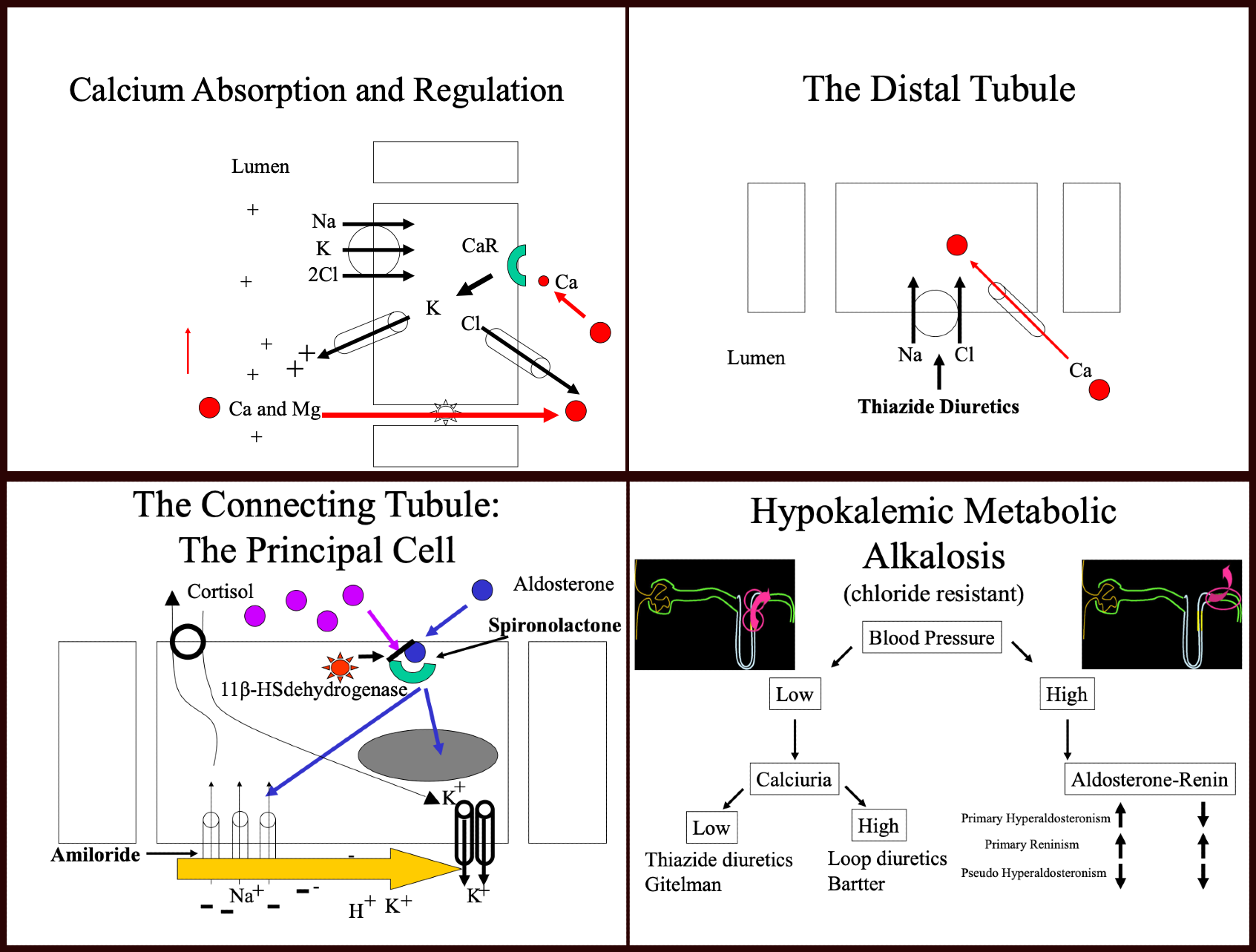
The diagnosis of Bartter syndrome results from the association of volume depletion, metabolic alkalosis, hypokalemia, and hypercalciuria. To explain the hypercalciuria, we need to review the calcium absorption in the thick ascending limb of Henle.
As most molecules, most of the calcium undergoes reabsorption in the proximal tubule.[6] A more fine-tuned regulation of calcium is made in the thick ascending limb through a paracellular pathway involving paracellin 1 (claudin 16).[7] An electrical gradient drives calcium and magnesium from a positively charged lumen towards the interstitium. A functional paracellular molecule is essential to this reabsorption. When the positive charges become decreased in the thick ascending limb, calcium is then retained in the lumen and lost in the urine, causing "hypercalciuria." This condition is a known feature of Bartter’s syndrome as well as loop diuretic use.
Much more common conditions can mimic these diseases:
- Loop Diuretic use or abuse
- Induced Vomiting
- The Distal Convoluted Tubule (DCT) [Diagram 2]
In the DCT, sodium is absorbed through a sodium chloride cotransporter (NCC).[8] It causes a salt-wasting syndrome described initially by Dr. Gitelman.[9] The syndrome features volume depletion, hypokalemia, metabolic alkalosis, and hypomagnesemia, as well as hypocalciuria. There are controversies as of the cause of hypocalciuria.[6] Initially thought to be a change of membrane potential by inhibition of NCC. This change in membrane potential would open the calcium channel (ECaC) present in the luminal side of the cell and so increase calcium absorption in this segment of the tubule.[10] Others have shown differently and have proved that it is, in fact, the reduced extracellular volume (ECV), which causes enhanced proximal tubule calcium reabsorption.[6][11]
More common conditions can mimic this disease:
- Thiazide use or abuse
- Induced vomiting
Earlier, this activity described salt-wasting conditions accompanied by hypokalemic metabolic alkalosis. It will now review salt retention conditions with hypokalemic metabolic alkalosis, which involve the principal cells of the connecting/collecting duct.
[Diagram 3]
Early experiments have shown that increased urine flow through the distal part of the nephron induces potassium losses. That finding led to the discovery that potassium excretion is proportionally related to the sodium content of this segment of the tubule. Further electrophysiological studies have shown that electrical charges in this segment were responsible for this effect.[12] Aggressive sodium reabsorption regulated by the renin-angiotensinogen aldosterone system (RAAS) leaves a negative charge in the lumen and so traps other positively charged ions, including protons and potassium. Potassium itself is highly regulated by aldosterone, which upon binding to its intracellular receptor, induces increased expression of epithelial sodium channels (ENaC) and potassium channels, which leads to sodium absorption and potassium excretion. Aldosterone secretion is regulated by renin secretion, itself controlled by the loss of volume in the distal convoluted tubule.
Potassium-sparing diuretics affect this segment of the nephron. Spironolactone binds and blocks the aldosterone receptor.
Amiloride blocks the sodium channel ENac.
Several syndromes affect the regulation of this system.
Primary Hyperaldosteronism
Dr. Conn initially described this syndrome and is due to excessive secretion of aldosterone by the zona glomerulosa of the adrenal gland. The histopathology will not be reviewed here. Excess of aldosterone binding to its receptor in the principal cells will, as mentioned, increase the expression of ENaC and potassium channels on the luminal side of those cells; this will dramatically increase sodium absorption and potassium excretion in the tubule. The excess of sodium absorption will leave a negatively charged lumen, which will augment potassium trapping in the lumen as well as protons leading to hypokalemia and metabolic alkalosis.
The treatment of this condition, in addition to blood pressure control, is the administration of spironolactone or eplerenone.
After the initial cases of Conn syndrome, multiple patients demonstrated similar features.
Primary Reninism
In 1967 a case of HTN due to a renin-secreting tumor was reported which led to the description by Dr. Conn of primary reninism.
The features of this syndrome are almost identical to primary hyperaldosteronism including volume retention, severe hypertension, and hypokalemic metabolic alkalosis with the only difference being that the culprit of all the clinical findings is an excess secretion of renin.
Evaluations of multiple patients with hypertension and hypokalemic metabolic alkalosis left many cases without an increase in aldosterone secretion to be called pseudohyperaldosteronism.
Liddle Syndrome
Initially described in 1963 by Dr. Liddle as a "familial renal disorder simulating primary hyperaldosteronism but with negligible aldosterone secretion" (Liddle G.W et al. 1963) was further determined to be a genetic mutation of the epithelial sodium channel inducing its overexpression at the luminal side of the principal cell causing unregulated absorption of sodium.[13] As in primary hyperaldosteronism, this causes hypertension, volume retention, and hypokalemic metabolic alkalosis. However, aldosterone and renin levels become decreased. In addition to treating hypertension, amiloride improves this condition.
Cushing Syndrome (Glucocorticoid excess)
Initially described by Dr. Cushing in 1932.[14] This disease causes the overstimulation of the corticoid receptor in the principal cells due to excess production of cortisol by the follicular layer of the adrenal gland. Upon binding to its receptor, cortisol has the same if not better affinity to the aldosterone receptor than aldosterone itself. There are many more molecules of cortisol than aldosterone in the serum, and this would overstimulate the receptor if it weren't for an enzyme (11-beta-HSdehydrogenase) which quickly converts cortisol to cortisone and renders it unable to bind the receptor. In the face of significant glucocorticoid excess, this enzyme is overwhelmed and leaves some of the hormones to bind the aldosterone receptor leading to an apparent aldosterone excess, which, in turn, induces volume retention, hypertension, and hypokalemia, and metabolic alkalosis but low aldosterone and low renin.
The use or abuse of steroids can mimic this.
Apparent Mineralocorticoid Excess
Even in the absence of glucocorticoid excess, several cases of apparent mineralocorticoid excess were described. It was found to be due to a genetic mutation of 11-beta-HSdehydrogenase which leaves the aldosterone receptor available to be bound by cortisol and leads to volume retention, hypertension, hypokalemia, and metabolic alkalosis.[15]
Of note, 11-beta-HSdehydrogenase can be inhibited by glycyrrhizic acid contained in natural licorice, and it is a well-known fact that the consumption of large amounts of licorice causes HTN with apparent mineralocorticoid excess.
Prognosis
Most of the genetic diseases inducing low blood pressure are usually detected in childhood with much more severe symptoms in the Bartter variety than in the Gitleman variety and so far has not affected the life span of those individuals. As for the high blood pressure category of diseases, it is clear that when a tumor is found (reninoma), leading to primary hyperaldosteronism and glucocorticoid excess, it requires surgical excision, and the genetic diseases can lead to complications of hypertension including, strokes, coronary artery disease, and end-stage kidney disease. Induced syndromes will have to be treated accordingly by changing the medication regimen or psychiatric evaluation but usually have a good prognosis.
Enhancing Healthcare Team Outcomes
Patient care is an interprofessional task at all times. The clinician is only aware of the patient's circumstances during their visit. Any overuse of medications and early prescription refills should be acknowledged and reported by the pharmacists to reduce abuse. Any other clinician or nurse seeing the patient and noticing changes, such as hypertension, dizziness, signs of vomiting, and dehydration, should discuss this with all the clinicians involved in the patient's care. The pharmacist should verify all dosing for potassium repletion as well as any diuretic therapy. These will be administered by nursing, who will also be the first to evaluate therapeutic effects and adverse events associated with medication treatment and report these to the clinician in charge of the case. Nursing must be vigilant in monitoring and report to the clinicians any status changes; they can also check patient compliance and answer patient questions. A collaborative interprofessional healthcare team approach is necessary for optimal patient outcomes, given the rarity and potential differentials for hypokalemic acidosis. [Level 5]