Continuing Education Activity
Orbital hypertelorism is defined as an increased distance between the orbits, with true lateral displacement of the orbits. Hypertelorism is associated with underlying craniosynostosis syndromes, median facial clefting, and some other genetic syndromes. Evaluation of hypertelorism includes precise measurement and a complete dysmorphology exam. Referral to a geneticist is indicated for patients with hypertelorism, and genetic testing is often indicated. Patients may benefit from evaluation by an interprofessional craniofacial team, and surgical reconstruction may be indicated.
Objectives:
- Identify the etiology of hypertelorism.
- Review the appropriate history, physical, and evaluation of hypertelorism.
- Summarize the management options available for hypertelorism.
- Describe interprofessional team strategies for improving care coordination and communication for patients with hypertelorism to improve outcomes.
Introduction
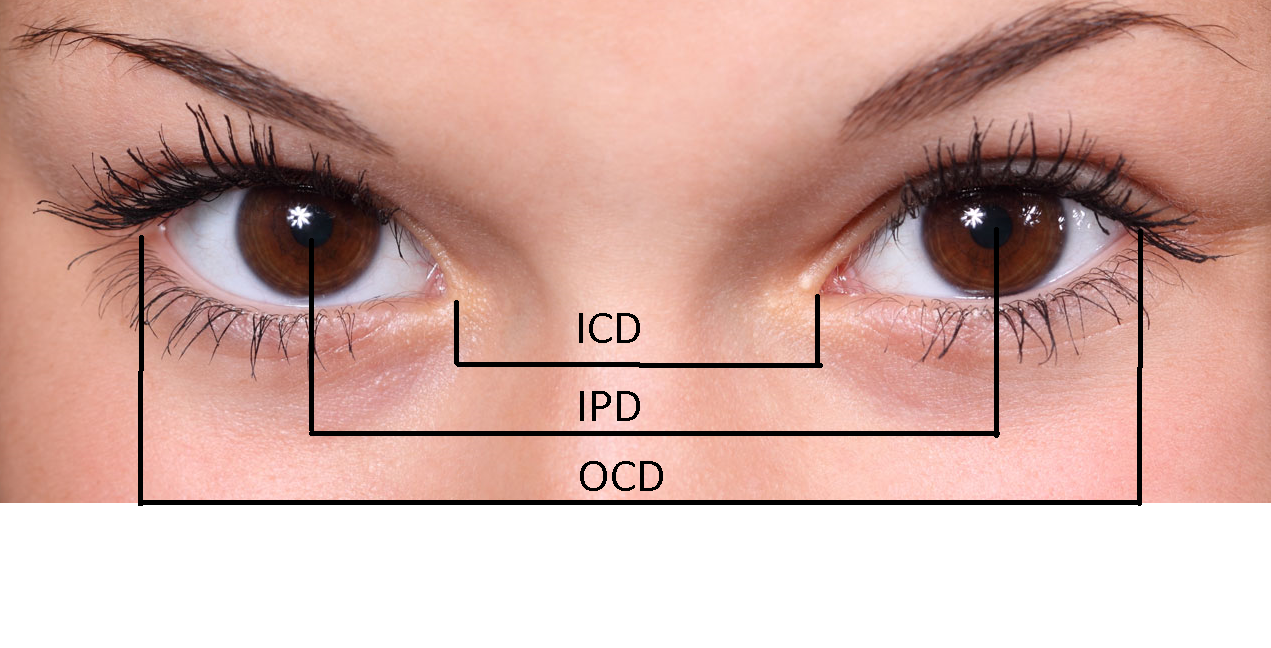
Orbital hypertelorism is defined as an increased distance between the orbits, with true lateral displacement of the orbits. Anthropometric measurements will yield increased inner canthal distance (ICD), increased outer canthal distance (OCD), and increased interpupillary distance (IPD). Increases in all three measurements above the 95th percentile on normative anthropometric values are observed in true orbital hypertelorism. Increased inner canthal distance alone is more properly described at telecanthus. For the sake of this article, hypertelorism will be used interchangeably with orbital hypertelorism. Additionally, this article will discuss true orbital hypertelorism, with a review of telecanthus in the section on differential diagnosis.
Etiology
Hypertelorism is thought to be caused by an alteration in embryological facial development during weeks 4-8 of development. The frontonasal prominence is the embryological precursor of the forehead and nose, and during normal development of the frontonasal prominence, there is lateral movement of the orbits followed by medial migration. If there is an arrest in the normal development of the frontonasal prominence, the primitive brain fills the space preventing the normal medial migration of the orbits and arresting them in a lateral position.[1]
Frank encephalocele and other masses in the frontonasal prominence can produce an obvious and extreme form of hypertelorism. Additional proposed mechanisms of hypertelorism include early ossification of the lesser wings of the sphenoid bone, preventing medial movement of the orbits and fixing them in the fetal position, and craniosynostosis syndromes where there is premature closure of cranial sutures preventing normal orbital migration and development.[2] In Apert syndrome, one of the most common craniosynostosis syndromes, hypertelorism is caused by the prolapse of the cribriform plate of the ethmoid bone, disturbing cranial base formation.[1]
Epidemiology
Hypertelorism is not a syndrome in and of itself, but rather it is a physical finding in many craniofacial syndromes. When thinking about the epidemiology of hypertelorism, it is helpful to consider the proposed embryological mechanisms. In the failure of the embryonic frontonasal prominence to develop normally, the patient will present with median facial clefting (also often referred to as frontonasal dysplasia). Alternatively, when dysplasia of bone development is the underlying contributive factor to hypertelorism, patients will present with craniosynostosis or cranial bone abnormalities.
Frontonasal dysplasia and or median facial clefting is rare, presenting in about 0.17% of all clefting presentations.[3] The epidemiology of all clefting is thought to be about 1 per 700 births.[4] The epidemiology of craniosynostosis is thought to be approximately 1 of 2,000 live births, though not all craniosynostosis syndromes have hypertelorism. The overall estimated incidence of hypertelorism is rare, with some sources estimating it to be about 1 in 20,000 births.[5]
There are additional genetic syndromes that may include hypertelorism as a feature, including but not limited to Bohring-Optiz Syndrome, Greig Cephalopolysyndactyly, Noonan Syndrome, Joubert Syndrome, Trisomy 18, 22q11 syndrome, Neurofibromatosis type 1, Aarskog syndrome, Cat-Eye Syndrome, CHARGE association, Loeys-Dietz syndrome, Wolf-Hirshorn Syndrome, Morquio Syndrome, and Hurler Syndrome. The specific epidemiology of each of these genetic syndromes is discussed separately in dedicated articles to each syndrome.
History and Physical
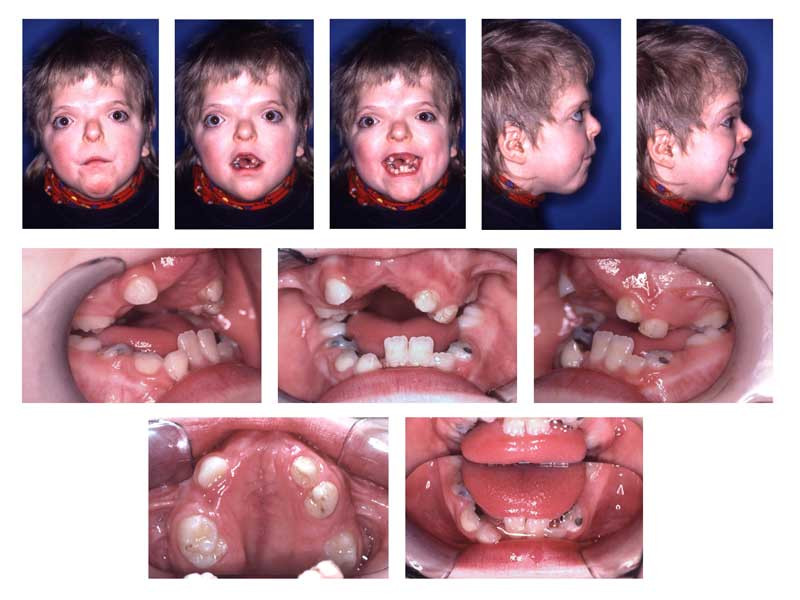
Isolated hypertelorism is rare. Thus it is important to evaluate a patient with hypertelorism for additional physical exam findings. Hypertelorism is typically noted at birth, and sometimes even before birth as fetal ultrasound becomes more sophisticated. Hypertelorism associated with medial clefting of the face and/or dysplasia of nasal capsule development is more likely to have other congenital anomalies. In one study evaluating prenatally diagnosed hypertelorism, karyotypic aneuploidy was the most common finding occurring in 82% of fetuses. All 11 fetuses showed additional anomalies of the nervous system (n = 10), skeleton (n = 6), heart (n = 5), kidneys (n = 5) and/or abdomen (n = 3).[6]
Hypertelorism associated with craniofacial synostosis may be syndromic or nonsyndromic. Of the craniosynostosis syndromes, Apert syndrome is associated with a sunken appearance of the midface, beaked nose, teeth crowding second to the underdeveloped maxilla, vision problems second to shallow eye sockets, mild to moderate intellectual disability, syndactyly or polydactyly, hyperhidrosis, and cleft palate.[7] Carpenter syndrome can present with a cloverleaf skull shape, low set ears, abnormal dentition, syndactyly or polydactyly, intellectual disability, umbilical hernia, obesity, congenital heart disease, hip and spine deformities, and genital abnormalities.[8] Crouzon syndrome can present with midface hypoplasia, beaked nose, dental abnormalities, hearing loss, and normal intelligence.[9]
Ultimately, the physical exam is variable, depending on the underlying etiology of hypertelorism. Thus, when hypertelorism is discovered as a physical finding, a full head to toe physical exam is necessary to identify additional dysmorphic features or congenital abnormalities.
Evaluation
The evaluation of hypertelorism entails specific and precise measurements. Bony inter-orbital measurements from radiograph or computed tomography are the most accurate measurements,[10] and are indeed important for surgical planning, however, are impractical for initial evaluation of a patient presenting with suspected hypertelorism. The precision with measurement can be attained when using proper technique and measuring distance between appropriate landmarks as detailed in the image of this article. Proper technique for measurement including has a patient sit comfortably on a chair in front of the examiner with the head at the same level as the head of the examiner, looking straight ahead. A small plastic transparent ruler measuring to the nearest millimeter is placed on the nasal bridge. Distances between the inner canthi, outer canthi, and palpebral fissures can be measured and recorded. Measurements can be compared to normative data, and hypertelorism documented for measurements above the 95th percentile in ICD, OCD, and IPD. Normative value tables are available online, and an abridged version is included here adapted from the standard normative values.[11][12]
Inner Canthal Distances in Centimeters by Age: Mean (2SD)
Premature newborn 1.6 (0.4)
Full-term newborn 2.0 (0.4)
1-6 months: 2.2 (0.5)
7-12 months: 2.5 (0.5)
13-18 months: 2.5 (0.6)
19-24 months: 2.5 (0.4)
25-30 months: 2.6 (0.6)
Outer Canthal Distances in Centimeters by Age: Mean (2SD)
Premature newborn 5.8 (0.7)
Full-term newborn 7.0 (0.8)
1-6 months: 7.5 (1.0)
7-12 months: 7.8 (1.4)
13-18 months: 8.5 (1.0)
19-24 months: 8.2 (1.0)
25-30 months: 8.6 (1.1)
Evaluation of a patient with hypertelorism, or pseudohypertelorism is not complete without a full head to toe for dysmorphology exam. Exam findings may offer clues to an underlying syndromic diagnosis. A complete dysmorphic exam may include the following evaluation, adapted from Smith.[13]
- Craniofacial
- Flat or prominent nasal bridge
- Small mandible
- Flat or prominent occiput
- Metopic ridge Low-set ears
- Large posterior fontanelle
- Malar hypoplasia
- Anteverted nose
- Ears
- Preauricular tags or sinus
- Large or small ears
- Asymmetric size
- Posterior rotation
- Lack of usual fold of helix
- Mouth
- Bifid uvula
- High-arched palate
- Wide alveolar ridges
- Large tongue
- Thin upper lip
- Flat philtrum
- Eyes
- Synophrys
- Epicanthal folds
- Hypo- and hypertelorism
- Ptosis
- Short palpebral fissures
- Upward slant to palpebral fissures
- Downward slant to palpebral fissures
- Skin/hair
- Low hairline
- Frontal upsweep/aberrant hair whorl
- Alopecia of scalp
- Extra posterior cervical skin
- A large capillary hemangioma (Other than on posterior neck)
- Café au lait spots
- Hypopigmented macules
- Chest
- Short sternum
- Depressed sternum
- Wide-set or high-located nipples
- Shield chest
- Abdominal/perineal
- Deep sacral dimple
- Diastasis recti (>3 cm) Aplasia cutis congenital
- Umbilical hernia
- Inguinal hernia
- Small testes
- Hypospadias
- Small or hypoplastic genitals
- Feet
- Syndactyly of toes
- Overlapping toes
- Wide gap ("sandal-gap") between toes
- Prominent heel
- Broad hallux
- Hallux valgus
- Hypoplastic nails
- Hands
- Single palmar crease
- Other unusual crease patterns
- Clinodactyly
- Duplication of nail
- Camptodactyly
- Partial cutaneous syndactyly
- Proximally placed thumb
- Broad thumb
- Duplication of thumbnail
- Small or dysplastic nails
- Overlapping fingers Long fingers
- Small or large hands
- Short metacarpals
Following classification is used based on inner canthal distance for hypertelorism in adults:
Tessier Classification
1st degree 30 – 34 mm ICD
2nd degree 35 – 40 mm ICD
3rd degree > 40 mm ICD
CT scan classification "Munro" based on the orientation of orbital walls :
Type I - parallel medial orbital walls more common
Type II - wedge-shaped posteriorly
Type III - oval widest dimension
Type IV - wedge-shaped anteriorly post to globe; difficult to correct
Treatment / Management
Surgery in hypertelorism is mainly for cosmetic purposes. It requires a major surgery that has to approach through intracranial as well as extracranial route. The pioneer in surgery for hypertelorism was Paul Tessier. It was he who demonstrated that orbits could be moved from there position without affecting vision. The aim of the surgery is to bring the two orbits together medially and correct any dystopia, correcting the nasal dorsal portion and removing excess of skin.
The timing of surgery is usually between 5 and 7 years. Before five years, the bone growth of the maxillary arch may be affected by inferior excision, which may affect dental formation. Moreover, the bones may not be stout enough to hold the osteotomies together. However, surgery has better predictability in adults; the psychological consequences of cosmetic blemish warrant early surgery. If there is associated craniosynostosis, it has to be corrected before one year of age independently.
Surgical options available are box osteotomy, facial bipartition, and U shaped osteotomy.[14]
Approaches depend upon the preoperative evaluation. The choice of procedure is determined by the presence of associated abnormalities, the morphology of the maxillary arch, the axis of the orbit, and the degree of hypertelorism.
Box osteotomy: Normal maxillary arch and dental occlusion, orbital axis are normal, mild to moderate hypertelorism.
Facial bipartition: narrow maxillary arch with incisors being higher than molars, the axis of the orbit is oblique, the nasal fossa is narrow, severe hypertelorism.
Differential Diagnosis
Psuedo-hypertelorism or the appearance of widely spaced eyes may occur when examining patients with features including a flat nasal bridge, epicanthic folds, exotropia, widely spaced eyebrows, or narrow palpebral fissures. Telecanthus is a term used to describe an increased inner canthal distance with preserved outer cantal distance. Telecanthus is often associated with prominent epicanthic folds. Disorders that feature telecanthus as a prominent finding include Down syndrome, Ehlers-Danlos syndrome, Klinefelter syndrome, fetal alcohol syndrome, Turner syndrome, Cri du Chat syndrome, and Waardenburg syndrome.[15]
Prognosis
Patients with mild, isolated hypertelorism generally have a good prognosis. Patients with hypertelorism associated with an underlying genetic syndrome may have mild to severe intellectual disability, along with morbidity associated with other congenital anatomical findings.
Complications
If surgical correction is pursued for hypertelorism, there are risks of complications as with any invasive surgery, including bleeding, CSF leak, and infection in the acute period. There is potential for late complications, including relapse of surgical correction second to midface soft tissue laxity and oculomotor deficiencies.[14]
Consultations
The finding of true orbital hypertelorism on the physical exam of infant warrants further investigation and consultation with a team of experts including geneticists, craniofacial surgeons, and ophthalmologists. Additional consultations may be warranted if a specific underlying genetic syndrome is identified.
Deterrence and Patient Education
The facial structures form in the first three months of pregnancy. During this developmental phase, the craniofacial anomalies begin. This may be due to the changes in genes or environmental factors. The evaluation and management requires a multidisciplinary approach between eye specialists and plastic surgeons and requires a long term follow up.
Enhancing Healthcare Team Outcomes
An interprofessional team that provides an integrated approach to evaluating hypertelorism can help achieve the best possible outcomes. A finding of ocular hypertelorism warrants further evaluation by a geneticist to evaluate the extent of hypertelorism, as well as to completing a head to toe dysmorphology exam to identify additional dysmorphic findings. Additional studies may be indicated, pending evaluation by an expert in dysmorphology. These may include CT head (to evaluate craniosynostosis), CT orbits, echocardiogram (to evaluated congenital heart disease), a renal ultrasound (to evaluate structural congenital malformations), skeletal survey, brain MRI (to evaluate underlying neuronal changes or evaluate for mass as indicated), retinal exam, audiology testing, sleep study, and others depending on the underlying etiology or genetic syndrome contributing to the finding of hypertelorism.