Continuing Education Activity
Graham-Little-Piccardi-Lasseur syndrome (GLPLS) is a rare variant of lichen planopilaris—a follicular form of lichen planus. Alongside frontal fibrosing alopecia and classic lichen planopilaris, GLPLS is 1 of 3 variants of lichen planopilaris. The etiology of GLPLS remains uncertain, but it is believed to involve a T-cell–mediated autoimmune mechanism. GLPLS symptoms typically begin with the appearance of hyperkeratotic papules on the trunk and extremities before alopecia develops. The syndrome is characterized by multifocal, patchy, cicatricial alopecia on the scalp, noncicatricial alopecia in the axillae and perineum, and follicular hyperkeratosis on the trunk and extremities.
Diagnosis relies on clinical presentation, histopathological analysis, and the exclusion of similar conditions. Treatment focuses on interrupting disease progression, relieving symptoms, and enhancing cosmesis through various modalities, including topical and systemic medications, surgical interventions, and patient support. Interventions must be tailored to each patient's needs and monitored for efficacy and safety. This activity reviews the histology, etiology, diagnosis, and management of GLPLS, as well as highlights the critical role of the interprofessional healthcare team in evaluating and treating this condition. This activity allows clinicians to treat GLPLS through tailored interventions by implementing appropriate treatment modalities and ensuring ongoing monitoring by an interprofessional healthcare team, which is crucial for improving patient care, prognosis, and quality of life.
Objectives:
Identify the clinical manifestations of Graham-Little-Piccardi-Lasseur syndrome, including hyperkeratotic papules on the trunk and extremities, and multifocal, patchy, cicatricial alopecia on the scalp.
Screen patients presenting with alopecia and hyperkeratosis for potential Graham-Little-Piccardi-Lasseur syndrome using appropriate diagnostic criteria and tests.
Select appropriate therapeutic interventions, including topical and systemic medications, surgical options, and supportive therapies, to ensure optimal patient outcomes.
Collaborate with an interprofessional healthcare team, including dermatologists, dermatopathologists, pharmacists, and psychologists, to provide comprehensive care for patients with Graham-Little-Piccardi-Lasseur syndrome.
Introduction
Graham-Little-Piccardi-Lasseur Syndrome Overview
Graham-Little-Piccardi-Lasseur syndrome (GLPLS) is a rare variant of lichen planopilaris—a follicular form of lichen planus. The condition was first described by Piccardi in 1913 and further characterized by Ernst Graham Little 2 years later in a mutual patient with Lasseur. [1]
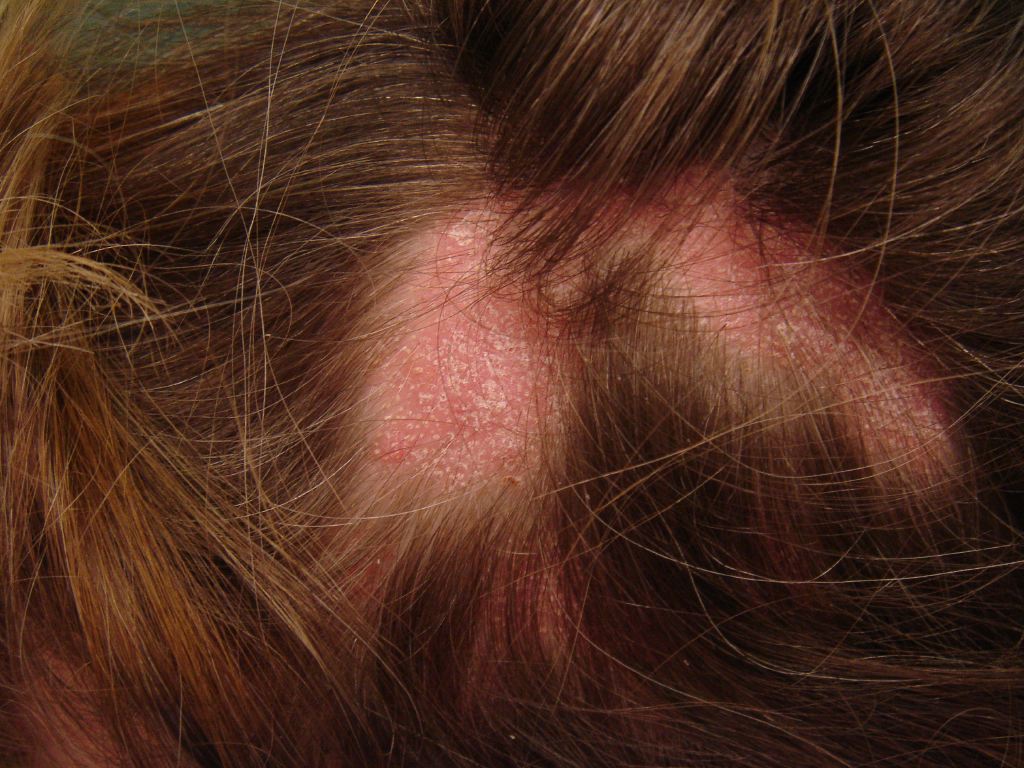
The 3 presentations of lichen planopilaris include GLPLS, frontal fibrosing alopecia, and classic lichen planopilaris.[2] Classic lichen planopilaris is a chronic inflammatory process manifesting as patchy progressive cicatricial scalp alopecia. Frontal fibrosing alopecia presents with band-like cicatricial alopecia on the scalp's frontal and temporal zones.[3][4] GLPLS is characterized by multifocal, patchy, cicatricial alopecia on the scalp, noncicatricial alopecia in the axillae and perineum, and follicular hyperkeratosis on the trunk and extremities.[5][6] GLPLS symptoms may occur in any order, often starting with hyperkeratotic papules on the trunk and extremities before alopecia develops in any location (see Image. Lichen Planopilaris).
GLPLS constitutes a small fraction of lichen planopilaris cases, predominantly affecting White women who are typically aged between 30 and 70. Correlations exist with various factors such as hepatitis B vaccination, androgen insensitivity syndrome, human leukocyte antigen (HLA)-DR-1 positivity (observed in a mother and daughter), vitamin A deficiency, hormonal dysfunction, and neuropsychological issues. Managing GLPLS focuses on immune response reduction to prevent follicular scarring, although reliable therapeutic regimens are still lacking. Research indicates that Janus kinase (JAK) inhibition with tofacitinib may normalize interferon (IFN)-mediated T-cell chemotaxis, potentially preventing infundibular inflammation. Despite theoretical advancements, healthcare professionals face challenges in achieving complete patient remission, underscoring the need for better preventive measures to mitigate long-term complications.
Hair Histology and the Hair Cycle
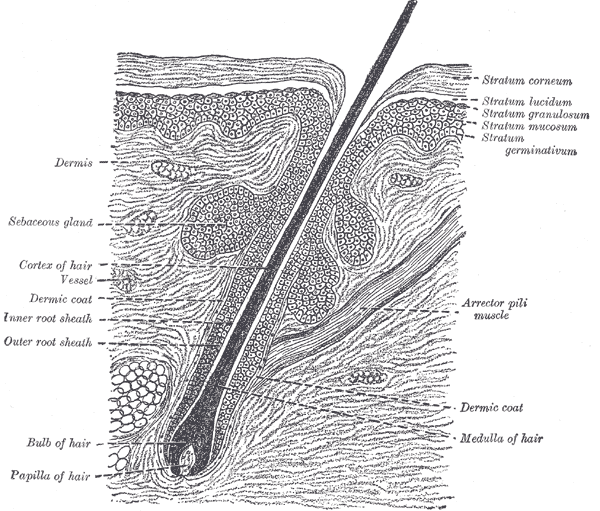
Hair follicles consist of upper (permanent) and lower (transitory) segments. The upper segment comprises the infundibulum, extending from the epidermis to the sebaceous duct, and the isthmus, which houses stem cells at the bulge region. The lower segment is divided into the trunk (stem) and the bulb, with the trunk extending from the insertion of the arrector pili muscle to the cornified part of the bulb (see Image. The Common Integument, Section of the Skin). The bulb becomes completely keratinized beyond this region, known as Adamson's fringe A, stretching to the hair follicle's base.
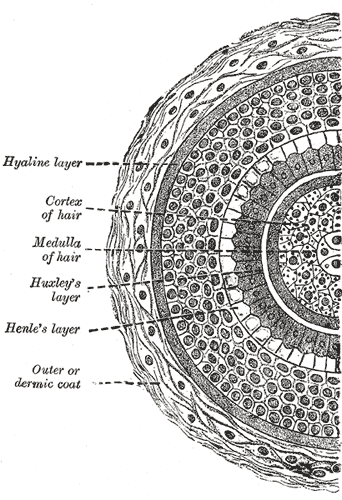
The follicular bulb houses the dermal papilla, providing blood supply and nutrients through a central capillary. The bulb also contains progenitor cells, dendritic melanocytes, and 3 internal hair sheath layers—Henle, Huxley, and cuticle—along with the external root sheath and vitreous membrane (basement membrane). The hair shaft has a cuticle, cortex, and medulla and becomes visible at higher levels of the follicle (see Image. Transverse Section of a Hair Follicle). The internal hair sheath merges into a single, fully keratinized stratum, while the external root sheath thickens with additional layers. Desquamation occurs in the upper trunk portions of the internal hair sheath, and keratinization takes place in the external root sheath. At the level of the infundibulum, the follicle undergoes keratinization similar to the epidermis, featuring a distinct granular layer and stratum corneum.
Hair can be classified into 3 types—lanugo, vellus, and terminal. Lanugo hair is fine and covers the fetus during development, typically disappearing after birth. Vellus hair is less than 1 cm long with a diameter of less than 0.03 mm, lacking a medulla, and its bulb is located in the upper dermis. Terminal hair is longer than 1 cm, with a diameter greater than 0.06 mm. This hair type possesses a cortex and a medulla, and its bulb is located in the subcutaneous tissue. Terminal hair is found in androgen-dependent (eg, scalp, beard, chest, axillae, and pubic region) and androgen-independent areas (eg, eyebrows and eyelashes). Androgens do not influence vellus hair.
The hair follicle's lower segment is involved in the hair growth cycle. About 85% to 100% of scalp hairs are typically in the anagen phase, which lasts 2 to 7 years. Around 1% of scalp hairs exist in the catagen phase—a period lasting 2 to 3 weeks. Matrix cell apoptosis and thinning of the lower segment occur during this period. In addition, the vitreous membrane and lower hair shaft thicken during this phase, surrounded solely by the external root sheath, creating a "club hair" appearance. Telogen is the resting phase, which involves 0% to 15% of scalp hairs, and about 100 days before the initiation of new hair growth and beginning of another cycle.[7][8]
Etiology
The etiology of GLPLS is an enigma, although most attribute GLPLS symptoms to a T-cell–mediated autoimmune process.[9] Mechanisms such as altered integrin expression and IFN-γ dysregulation, present in lichen planopilaris, likely contribute to GLPLS. Inflammation and altered integrin expression may lead to cicatricial alopecia, with affected follicular roots showing positive anagen hair pull tests.[10][11] Rodriguez-Bayona et al reported strong autoimmune reactivity to a centromere-specific protein, inner centromere protein or "INCENP," suggesting mitotic cellular deficits. Although further investigation is needed on the prevalence of INCENP autoantibodies in GLPLS, this finding marks the first GLPLS-linked antibody.[12] Familial GLPLS across 3 generations has also been reported recently.[13]
Epidemiology
GLPLS mainly affects White women, with an average onset age ranging from 30 to 70.[14] This lichen planopilaris variant comprises a small portion of the overall cases. In a study involving 80 individuals with lichen planopilaris, only 1 case was reported consistent with GLPLS. The condition correlates with various factors, including hepatitis B vaccination, androgen insensitivity syndrome, HLA DR-1 positivity in a mother and daughter, vitamin A deficiency, hormonal dysfunction, and neuropsychological issues.[15]
Pathophysiology
Damage to the pilosebaceous unit is crucial in GLPLS pathogenesis. Lesional skin exhibits increased IFN-γ–induced chemokines, recruiting cytotoxic T cells to the follicular infundibulum.[16] Dysregulation of the IFN-γ signaling pathway increases the expression of major histocompatibility complex classes 1 and 2, enhancing antigen presentation to T cells. Damage to the chromosomal passenger protein INCENP impedes microtubule binding, causing mislocalization and dysfunction of chromosome passenger complexes. Consequently, chromosomal segregation, central spindle formation, and cytoplasmic allocation are impaired.[17][18] These alterations create metaphase abnormalities that reduce overall mitotic function.
Histopathology
Histopathological examination of cicatricial alopecia areas in GLPLS reveals a dilated follicular lumen superior to the sebaceous gland with a dense hyperkeratotic plug in the lower portion of the infundibulum. The nearby epithelium peripheral to the follicular infundibulum shows hypergranulosis, vacuolar keratinocyte degradation, and a lymphocytic infiltrate obscuring the dermal-epidermal junction, along with increased dense collagen bundles at the follicular base. Examination of hyperkeratotic papules on the trunk and extremities shows expanded keratin-filled follicular openings, vacuolar degradation of adjacent keratinocytes, and a lymphohistiocytic infiltrate at the dermal-epidermal junction (see Image. Histopathology of Lichen Planopilaris).[19]
History and Physical
History
A thorough clinical evaluation is key to diagnosing GLPLS. On history, patients with this condition typically present with a unique hair loss pattern characterized by multifocal, patchy alopecia with hair follicle scarring (cicatricial). Patients may also exhibit noncicatricial alopecia in the axillae and perineum. Follicular hyperkeratosis is often reported on the trunk and extremities. Symptoms are gradual in onset, with hyperkeratotic papules often appearing before hair loss. These papules may evolve into hyperkeratotic plaques over time. Information that may help assess for secondary alopecia causes includes the amount of hair loss, the presence of psychosocial stress, or endocrinological abnormalities.
Physical Examination
Physical examination should include a full-body skin check, noting the multifocal, patchy cicatricial scalp hair loss and nonscarring perineal and axillary alopecia. Hyperkeratotic papules with surrounding perifollicular erythema on the trunk and extremities are key features of GLPLS. An anagen hair pull test is often positive in GLPLS because the hair follicles are weakly anchored in affected individuals. Dermoscopy may reveal perifollicular erythema and scaling in alopecic areas (see Image. Dermoscopic and Clinical Findings in Lichen Planopilaris).
Evaluation
Skin biopsy and physical examination findings serve as the primary diagnostic methods for GLPLS. Histological analysis of cicatricial areas and hyperkeratotic papules reveals lichen planopilaris features, indicating perifollicular inflammation leading to pilosebaceous destruction. Late-stage GLPLS may involve arrector pili muscle corruption. Clinical evaluation typically shows erythematous follicular papules varying in size from 1 to 4 mm, nonscarring alopecia in the perineum and axillae, and patchy cicatricial scalp alopecia. Hyperkeratotic papules commonly appear on the trunk, thighs, inner and outer arms, and wrists, with or without pruritus. These papules may precede the onset of scalp alopecia, but the order of symptoms can vary.[20]
Treatment / Management
Managing GLPLS remains elusive due to the lack of a dominant therapeutic regimen. Treatment focuses on mitigating the immunological response before follicular scarring occurs. Novel research suggests that JAK inhibition with tofacitinib normalizes IFN-mediated T-cell chemotaxis, preventing infundibular inflammation. A case series of 8 lichen planopilaris patients reported that a twice-daily 5-mg dose of tofacitinib resulted in a 30% to 94% improvement in disease severity.
Zegarska et al described a patient treated with prednisone 30 mg for 2 weeks, followed by a reduction to 20 mg, alongside psoralen and ultraviolet light A at 12 J (joules). This regimen moderately improved follicular hyperkeratosis on the trunk and extremities.[21] While the follicular papules were reduced, cicatricial scalp alopecia and noncicatricial axillary and perineal hair loss did not diminish.[22]
Other therapies used to manage GLPLS include intralesional glucocorticoids, topical glucocorticoids, systemic glucocorticoids, cyclosporine, tacrolimus ointment, and systemic retinoids. These treatments typically reduce perifollicular erythema and inflammation but may have a limited impact on scalp, perineal, and axillary hair loss. Recent reports have documented successful treatment of GLPLS using narrow-band UVB phototherapy.[23]
Differential Diagnosis
The differential diagnoses of GLPLS include:
- Classical lichen planopilaris
- Pseudopelade of Brocq [24]
- Frontal fibrosing alopecia
- Lichen spinulosus
- Androgen insensitivity syndrome
- Discoid lupus erythematosus [25]
- Secondary syphilis [26]
- Addison disease [27]
These conditions often manifest with skin lesions and scalp conditions resembling GLPLS symptoms. A thorough clinical investigation and appropriate diagnostic testing can guide management strategies.
Prognosis
The course of GLPLS is often drawn out, and while it may improve with treatment, complete resolution of alopecic symptoms is rare with current modalities. Data on the average disease duration of GLPLS are limited, although some cases persist for over 20 years. Zegarska et al reported durations ranging from 6 months to 10 years.[22] Adequate medical management may significantly improve the prognosis, particularly in reducing hyperkeratotic papules on the trunk and extremities.
Complications
GLPLS can cause considerable psychosocial distress due to its significant impact on a person's appearance. However, the condition does not exhibit systemic manifestations. Further research on INCENP's role, especially in overall mitotic function, may uncover associated morbidities, although none have been identified to date. The lasting complication of GLPLS is follicular scarring resulting from infundibular inflammation, which permanently damages pilosebaceous units, particularly in the scalp.
Deterrence and Patient Education
Patients with GLPLS require comprehensive explanations about prognosis and treatment to establish realistic expectations. Many patients opt out of treatment because current therapies often do not address the condition's alopecic aspects. Although treatments cannot reverse cicatricial damage, early intervention may slow the progression of noncicatricial alopecia, cicatricial scalp alopecia, and hyperkeratotic papule formation. Clarifying treatment goals can enhance compliance and foster stronger patient-physician relationships.
Pearls and Other Issues
GLPLS is a complex clinical entity characterized by multifocal, patchy cicatricial scalp alopecia, noncicatricial alopecia in areas such as the axillae and perineum, and hyperkeratotic papules on the trunk and extremities. Diagnosis relies on integrating clinical presentation, histopathological findings, and exclusion of similar conditions. Treatment remains challenging, focusing on immunomodulatory agents to reduce inflammation and disease progression. While therapies may not reverse existing cicatricial damage, early intervention can slow further hair loss and papule formation. Educating patients about treatment goals promotes compliance and supports strong patient-physician relationships.
Enhancing Healthcare Team Outcomes
An interprofessional approach involving various healthcare specialties, such as dermatologists, dermatopathologists, pharmacists, hair-loss support groups, and psychologists, is essential for the comprehensive evaluation and management of GLPLS. These multidisciplinary healthcare professionals are critical in accurately diagnosing the condition, treatment planning, and maintaining long-term care. In addition to medical care, trained healthcare professionals and alopecia support groups offer vital psychosocial support, thereby fostering a sense of belonging and reducing stress associated with a GLPLS diagnosis.[28]