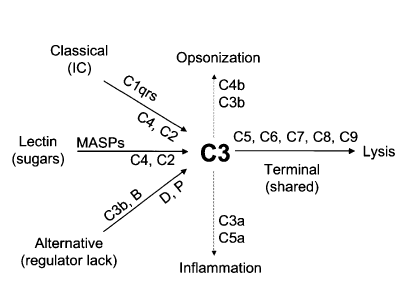
[2]
Schröder-Braunstein J, Kirschfink M. Complement deficiencies and dysregulation: Pathophysiological consequences, modern analysis, and clinical management. Molecular immunology. 2019 Oct:114():299-311. doi: 10.1016/j.molimm.2019.08.002. Epub 2019 Aug 14
[PubMed PMID: 31421540]
[3]
Blanco P, Pellegrin JL, Moreau JF, Viallard JF. [Pathophysiology of systemic lupus erythematosus]. Presse medicale (Paris, France : 1983). 2007 May:36(5 Pt 2):825-34
[PubMed PMID: 17449371]
[4]
Contin-Bordes C, Lazaro E, Pellegrin JL, Viallard JF, Moreau JF, Blanco P. [Systemic lupus erythematosus: from pathophysiology to treatment]. La Revue de medecine interne. 2009 Dec:30(12 Suppl):H9-13
[PubMed PMID: 19995652]
[5]
Skattum L, van Deuren M, van der Poll T, Truedsson L. Complement deficiency states and associated infections. Molecular immunology. 2011 Aug:48(14):1643-55. doi: 10.1016/j.molimm.2011.05.001. Epub 2011 May 31
[PubMed PMID: 21624663]
[6]
Lee JX, Yusin JS, Randhawa I. Properdin deficiency-associated bronchiectasis. Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology. 2014 Jun:112(6):557-9. doi: 10.1016/j.anai.2014.04.003. Epub 2014 May 1
[PubMed PMID: 24793003]
[7]
Jönsson G, Hansson C, Mellhammar L, Gullstrand B, Bengtsson AA, Sahl C, Skattum L. Increased serum bactericidal activity of autologous serum in C2 deficiency after vaccination against Haemophilus influenzae type b, and further support for an MBL-dependent C2 bypass mechanism. Vaccine. 2021 Feb 22:39(8):1297-1302. doi: 10.1016/j.vaccine.2021.01.007. Epub 2021 Jan 25
[PubMed PMID: 33509693]
[8]
Miller EC, Chase NM, Densen P, Hintermeyer MK, Casper JT, Atkinson JP. Autoantibody stabilization of the classical pathway C3 convertase leading to C3 deficiency and Neisserial sepsis: C4 nephritic factor revisited. Clinical immunology (Orlando, Fla.). 2012 Dec:145(3):241-50. doi: 10.1016/j.clim.2012.09.007. Epub 2012 Sep 28
[PubMed PMID: 23117396]
[9]
Agarwal S, Ferreira VP, Cortes C, Pangburn MK, Rice PA, Ram S. An evaluation of the role of properdin in alternative pathway activation on Neisseria meningitidis and Neisseria gonorrhoeae. Journal of immunology (Baltimore, Md. : 1950). 2010 Jul 1:185(1):507-16. doi: 10.4049/jimmunol.0903598. Epub 2010 Jun 7
[PubMed PMID: 20530262]
[10]
Bryan AR, Wu EY. Complement deficiencies in systemic lupus erythematosus. Current allergy and asthma reports. 2014 Jul:14(7):448. doi: 10.1007/s11882-014-0448-2. Epub
[PubMed PMID: 24816552]
[11]
Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Molecular immunology. 2014 Oct:61(2):110-7. doi: 10.1016/j.molimm.2014.06.030. Epub 2014 Jul 15
[PubMed PMID: 25037634]
Level 3 (low-level) evidence
[12]
Gigli I. Immunochemistry and immunobiology of the complement system. The Journal of investigative dermatology. 1976 Sep:67(3):346-53
[PubMed PMID: 787430]
[13]
Jlajla H, Dehman F, Jallouli M, Khedher R, Ayadi I, Zerzeri Y, Laadhar L, Sfar I, Mahfoudh A, Gorgi Y, Cheour E, Zouaghi K, Gargah T, Kallel Sellami M. Molecular basis of complement factor I deficiency in Tunisian atypical haemolytic and uraemic syndrome patients. Nephrology (Carlton, Vic.). 2019 Mar:24(3):357-364. doi: 10.1111/nep.13217. Epub
[PubMed PMID: 29292855]
[14]
Roumenina LT, Sène D, Radanova M, Blouin J, Halbwachs-Mecarelli L, Dragon-Durey MA, Fridman WH, Fremeaux-Bacchi V. Functional complement C1q abnormality leads to impaired immune complexes and apoptotic cell clearance. Journal of immunology (Baltimore, Md. : 1950). 2011 Oct 15:187(8):4369-73. doi: 10.4049/jimmunol.1101749. Epub 2011 Sep 19
[PubMed PMID: 21930969]
[15]
Grumach AS, Goudouris E, Dortas Junior S, Marcelino FC, Alonso MLO, Martins RO, Arpon MA, Valle SOR. COVID-19 affecting hereditary angioedema patients with and without C1 inhibitor deficiency. The journal of allergy and clinical immunology. In practice. 2021 Jan:9(1):508-510. doi: 10.1016/j.jaip.2020.11.042. Epub 2020 Nov 30
[PubMed PMID: 33271349]
[17]
Sonea MJ, Moroz BE, Reece ER. Leiner's disease associated with diminished third component of complement. Pediatric dermatology. 1987 Aug:4(2):105-7
[PubMed PMID: 2958789]
[18]
Goodyear HM, Harper JI. Leiner's disease associated with metabolic acidosis. Clinical and experimental dermatology. 1989 Sep:14(5):364-6
[PubMed PMID: 2532987]
[19]
Degn SE, Jensenius JC, Thiel S. Disease-causing mutations in genes of the complement system. American journal of human genetics. 2011 Jun 10:88(6):689-705. doi: 10.1016/j.ajhg.2011.05.011. Epub
[PubMed PMID: 21664996]
[20]
van Schaarenburg RA, Magro-Checa C, Bakker JA, Teng YK, Bajema IM, Huizinga TW, Steup-Beekman GM, Trouw LA. C1q Deficiency and Neuropsychiatric Systemic Lupus Erythematosus. Frontiers in immunology. 2016:7():647. doi: 10.3389/fimmu.2016.00647. Epub 2016 Dec 27
[PubMed PMID: 28082982]
[21]
Peerschke EI, Yin W, Alpert DR, Roubey RA, Salmon JE, Ghebrehiwet B. Serum complement activation on heterologous platelets is associated with arterial thrombosis in patients with systemic lupus erythematosus and antiphospholipid antibodies. Lupus. 2009 May:18(6):530-8. doi: 10.1177/0961203308099974. Epub
[PubMed PMID: 19395455]
[22]
Lewis LA, Ram S. Meningococcal disease and the complement system. Virulence. 2014 Jan 1:5(1):98-126. doi: 10.4161/viru.26515. Epub 2013 Oct 8
[PubMed PMID: 24104403]
[23]
Audemard-Verger A, Descloux E, Ponard D, Deroux A, Fantin B, Fieschi C, John M, Bouldouyre A, Karkowsi L, Moulis G, Auvinet H, Valla F, Lechiche C, Davido B, Martinot M, Biron C, Lucht F, Asseray N, Froissart A, Buzelé R, Perlat A, Boutboul D, Fremeaux-Bacchi V, Isnard S, Bienvenu B. Infections Revealing Complement Deficiency in Adults: A French Nationwide Study Enrolling 41 Patients. Medicine. 2016 May:95(19):e3548. doi: 10.1097/MD.0000000000003548. Epub
[PubMed PMID: 27175654]
[24]
Ekdahl KN, Persson B, Mohlin C, Sandholm K, Skattum L, Nilsson B. Interpretation of Serological Complement Biomarkers in Disease. Frontiers in immunology. 2018:9():2237. doi: 10.3389/fimmu.2018.02237. Epub 2018 Oct 24
[PubMed PMID: 30405598]
[25]
Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clinical microbiology reviews. 2010 Oct:23(4):740-80. doi: 10.1128/CMR.00048-09. Epub
[PubMed PMID: 20930072]
[26]
Sobh A, Bonilla FA. Vaccination in Primary Immunodeficiency Disorders. The journal of allergy and clinical immunology. In practice. 2016 Nov-Dec:4(6):1066-1075. doi: 10.1016/j.jaip.2016.09.012. Epub
[PubMed PMID: 27836056]
[27]
Műzes G, Sipos F. [Primary immunodeficiency and autoimmune diseases]. Orvosi hetilap. 2018 Jun:159(23):908-918. doi: 10.1556/650.2018.31064. Epub
[PubMed PMID: 29860882]
[28]
Dellepiane RM,Dell'Era L,Pavesi P,Macor P,Giordano M,De Maso L,Pietrogrande MC,Cugno M, Invasive meningococcal disease in three siblings with hereditary deficiency of the 8(th) component of complement: evidence for the importance of an early diagnosis. Orphanet journal of rare diseases. 2016 May 17
[PubMed PMID: 27183977]
[29]
Dellepiane RM, Baselli LA, Cazzaniga M, Lougaris V, Macor P, Giordano M, Gualtierotti R, Cugno M. Hereditary Deficiency of the Second Component of Complement: Early Diagnosis and 21-Year Follow-Up of a Family. Medicina (Kaunas, Lithuania). 2020 Mar 10:56(3):. doi: 10.3390/medicina56030120. Epub 2020 Mar 10
[PubMed PMID: 32164349]