Continuing Education Activity
Cheilitis granulomatosa (CG) is a rare granulomatous disorder characterized by a recurrent firm swelling of one or both lips. This is called the cheilitis granulomatosa of Miescher (CGM) when it occurs in isolation. It is called Melkersson-Rosenthal syndrome (MRS) if the classical triad of recurrent or persistent orofacial edema, plicated or fissured tongue (lingua plicata), and relapsing peripheral facial nerve paralysis is present. Cheilitis granulomatosa has many differential diagnoses that merit ruling out. This activity describes the evaluation and management of cheilitis granulomatosa and highlights the role of the interprofessional team in improving care for affected patients.
Objectives:
- Describe the etiology of cheilitis granulomatosa.
- Explain how cheilitis granulomatosa is diagnosed.
- Review the therapeutic options available for cheilitis granulomatosa.
- Summarize a well-coordinated interprofessional team approach to enhance the care of patients affected by cheilitis granulomatosa.
Introduction
Cheilitis granulomatosa (CG) is a rare chronic disease characterized by a recurrent firm swelling of one or both lips, and, histologically, by a granulomatous infiltrate. An isolated granulomatous machrochelitis defines the cheilitis granulomatosa of Miescher (CGM).[1][2] The Melkersson-Rosenthal syndrome (MRS) is characterized, in its complete form, by a classical triad of symptoms: recurrent or persistent orofacial edema (facial and lip edemas), plicated or fissured tongue (lingua plicata), and relapsing peripheral facial nerve paralysis. Most of the cases of MRS present with partial symptoms. CGM is the most common monosymptomatic form of MRS.[3][4] Other diagnoses that are also a part of the orofacial granulomatosis group must be eliminated. The orofacial granulomatosis, first individualized in 1985, by Wiesenfeld et al., is a syndrome grouping noninfectious and nonnecrotizing granulomatous involvement of the lips, oral cavity, and face, in addition to the CG, sarcoidosis, and Crohn's disease.[5]
Etiology
The etiology of CG is still unknown. Several etiologies of different orders have been proposed including genetic, inflammatory, allergic, and microbial. There is not a clear etiological mechanism.[6][7]
The postulated etiological factors include:
- Genetic predisposition: Genetic origin has been suggested considering some cases with involvement of other family members; however, it remains unproven, and no HLA association has been found in patients with CG compared to the general population.
- Immunologic factors: CG is a chronic inflammatory condition characterized by a predominant T helper 1-mediated immune response. The main anomaly is most likely a local alteration of innate immunity of lip mucosa in response to various antigens whose insufficient purification leads to a persistent granulomatous reaction.
- Allergic factors: Allergy to dental materials, foodstuffs, and food additives, has been suspected, but the causal relationship is not well established.
- Microbial factors: Several microbial agents have been incriminated, as eventual factors of deregulation of the immune response. Studies have included especially Mycobacterium tuberculosis and paratuberculosis, Borrelia burgdorferi, Saccharomyces cerevisiae, and Candida albicans. Nevertheless, with conflicting results, the role of these microbes in the pathogenesis of CG remains uncertain.
- Hypersensitivity to ultraviolet (UV)-B radiation.
- As part of the spectrum of Crohn's disease: Although early studies suggested that CG may represent an extra-intestinal variant of Crohn's disease with overt gastrointestinal disease developing much later, the fact that CG is found in less than 1% of patients with Crohn's disease cannot be ignored.
Epidemiology
CG is a rare disease of undefined incidence and prevalence. One study estimated incidence to be 0.08% of the general population. It may have an onset in all age groups but is most commonly seen in adults with peak incidence reported between 20 to 40 years of age. It rarely affects children. However, in two recently published articles, 30 cases in pediatric patients have been described. Most of the literature suggests an equal sex distribution; however, some authors reported that it is more frequently reported in females. Rare familial cases have also been reported.
Histopathology
Histological examination is not necessary for the diagnosis of the complete form of MRS, which can be easily established based on clinical findings. Nevertheless, the histological analysis is necessary for incomplete forms, including CGM, especially to eliminate the differential diagnoses of sarcoidosis, Crohn's disease, and other granulomatous disorders including cutaneous tuberculosis, leishmaniasis, and leprosy, especially in endemic areas. The histological characteristics of CG (including CGM and MRS) are granulomatous infiltrate constituted by epithelioid cells and multinucleate giant cells, without caseous necrosis, associated with some degree of lymphedema and fibrosis. These histopathological features are not constant, and their absence should not formally exclude the diagnosis of CG.
History and Physical
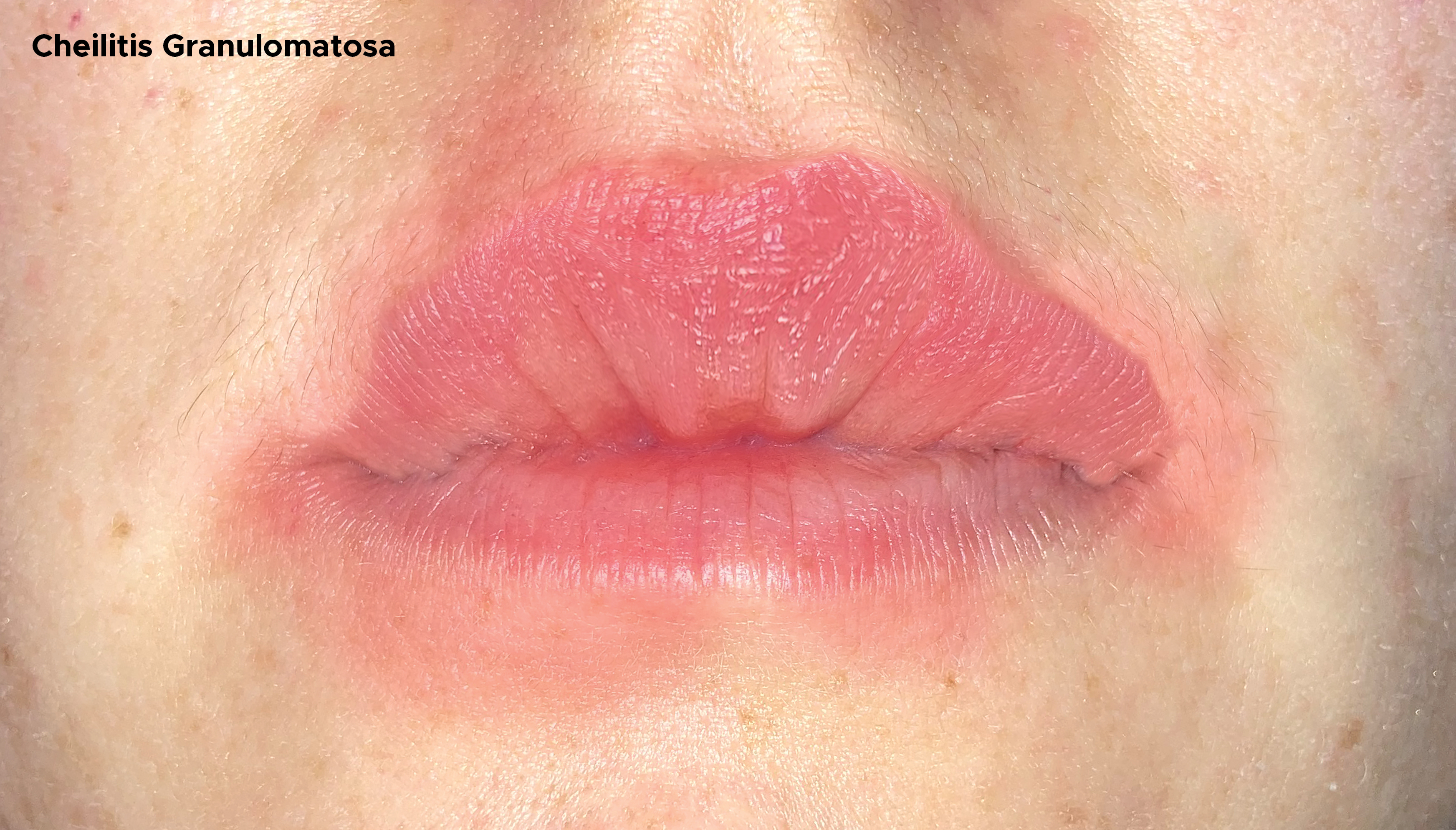
MRS consists of a triad of recurrent or persistent orofacial (lips or face) swelling, peripheral facial nerve paralysis, and fissural or scrotal tongue.[8][9] Labial swelling is initially characterized by recurrent edematous swellings mimicking angioedema. Subsequently, after many episodes, it becomes persistent and indurated. It is a firm non-erythematous non-painful swelling affecting one or both lips. Rarely, the labial swelling can cause difficulties in speech or may result in drooling.
The complete classical triad is uncommon and may be observed in one-fourth to one-third of the patients. In the complete form of MRS, all symptoms rarely appear simultaneously, and long intervals between the occurrence of paralysis and the first swelling have been reported. The disease evolves by flare-ups; nevertheless, permanent edema of the lips, and sometimes on the face, can gradually be observed. The oligosymptomatic and monosymptomatic forms are most frequent at the beginning of the disease, and they represent 40% of the cases. CGM or Miescher syndrome corresponds to an incomplete form of MRS in 28% of the cases. Thus, it represents the most common form of MRS. It generally affects the upper lip and less frequently the lower lip. In addition, changes in the buccal, palatal, sublingual, and gingival mucosa are anecdotally reported.
Oral manifestations of sarcoidosis and Crohn's disease may mimic those of CGM or MRS.[10][11][12]
Evaluation
CG is mainly a clinical diagnosis. Histopathology should be considered and, if positive, may be extremely helpful in diagnostic confirmation. Investigations to rule out sarcoidosis (high-resolution computed tomography scan of chest, serum angiotensin-converting enzyme levels) and purified protein derivative (PPD) testing may be done as per the clinical scenario. Since the period between the onset of lip swelling and the appearance of typical symptoms of Crohn's disease is highly variable and unpredictable, the role of investigations like colonoscopy or gastroduodenoscopy with biopsies in patients without gastrointestinal symptoms remains controversial.
Treatment / Management
The rarity of this disease explains the empiricism of therapeutic proposals and the absence of controlled studies. An effective medical treatment is not available at present. The proposed symptomatic treatments are simply intended to avoid or space recurrences, particularly in the edematous stage. The treatment aims to relieve these patients and to improve their quality of life, often very disturbed by the unsightly and distressing nature of macrocheilitis and oro-facial edema. The spontaneous disappearance of the disease is rare but has been reported.
Corticosteroid therapy is a classic treatment for CG. It may be administered locally, topically, or intralesionally, and at times systemic corticosteroid for short courses (prednisolone: 0.3 to 0.7 mg/kg/day; 25 to 50 mg/day) may be tried. Other treatments have also been tried for their anti-inflammatory or immunomodulatory effects such as topical tacrolimus, or oral clofazimine, thalidomide, dapsone, and doxycycline, with inconsistent results.
Finally, immunosuppressive treatments such as mycophenolate mofetil and azathioprine, as well as inhibitors of tumor necrosis factor-alpha (TNF-alpha), have been tried, alone or in combination, in isolated cases and small series with inconsistently positive results.
Fumaric acid esters have given good results in 50 % of the cases (who were refractory to other modalities).
The cheiloplasty reduction is possible when the lesions are fixed and not evolving.
Differential Diagnosis
The list of differentials of lip swelling are many, including:
- Acquired angioedema due to C1 deficiency
- Hereditary angioedema
- Glandular cheilitis
- Dental abscess
- Crohn's disease
- Cutaneous manifestation of various granulomatous disorders including sarcoidosis, tuberculosis, leishmaniasis, leprosy, Wegener granulomatosis
The histopathology of angioedema is not granulomatous. Moreover, some conditions often have a family history and episodic bouts of abdominal pain. A dental abscess can be ruled out with a thorough dental examination and swabs. Differentiation from other granulomatous disorders has been discussed above.
Prognosis
Owing to the scarcity of literature, poor understanding of the pathophysiology, and modest response to therapy, the overall prognosis of the CG spectrum or OFG is not encouraging. The monosymptomatic form, i.e., CGM, may resolve with the treatments mentioned above. MRS has the poorest prognosis with no published evidence on the response of neurological complications of MRS to treatment.
Complications
As such, CGM is a benign disorder. But since CG may be a part of MRS, the occurrence of facial palsy later is a distinct possibility. If CG in an individual is representative of evolving Crohn's disease, then complications of inflammatory bowel disease may occur.
Deterrence and Patient Education
The patient should be counseled about the possibility of lip swelling representing CGM alone but also cautioned to report if facial paresis develops, as it may be suggestive of MRS. Careful counseling and explanation about the availability of limited therapeutic options are important.
Pearls and Other Issues
CG is a rare disorder with a difficult diagnosis, characterized by recurrent or persistent idiopathic swelling of one or both lips due to granulomatous inflammation. When isolated, it defines Miescher syndrome. However, other clinical signs may be associated, such as facial nerve paralysis and fissured tongue, which complete the triad of MRS. The etiopathogenesis and the etiology of CG are still obscure, and the treatment, therefore, remains uncertain. In addition to the unsightly nature of this condition and the significant psychological impact, there may be local complications like excessive lacrimation, dry eyes, disorders of salivary secretion, and dysgeusia. Possible familial cases should be searched. Further studies are needed to explain the pathophysiology and find other, more effective therapeutic alternatives.
Enhancing Healthcare Team Outcomes
As the patient may present to a primary care clinician/surgeon as well as a specialist, including a dermatologist, dentist, or otolaryngologist, all the care providers should be cognizant about the condition and its associations. As the majority of the work on this condition has been done by dermatologists, other clinicians should not delay referring the patient early. Similarly, meticulous examination for facial paresis may need the otolaryngologist or even a neurologist. Thus, interprofessional coordination is vital in the management of this condition. The rarity of this disease explains the empiricism of therapeutic proposals and the absence of controlled studies. An effective medical treatment is not available at present. The proposed symptomatic treatments are simply intended to avoid or space recurrences, particularly in the edematous stage. The treatment aims to relieve these patients and to improve their quality of life, often very disturbed by the unsightly and distressing nature of macrocheilitis and oro-facial edema. The spontaneous disappearance of the disease is rare but has been reported.