Continuing Education Activity
Incontinentia pigmenti is an X-linked dominant genodermatosis. The disease is caused by a mutation of the IKBKG gene. This rare disorder presents in the first few weeks of life. Skin lesions are typical of this disease. The eye, teeth, and central nervous system are also involved. Due to multisystemic involvement, it must be promptly diagnosed and treated. Genetic counseling plays an important part in the management. This activity reviews the evaluation and diagnosis of incontinentia pigmenti and highlights the role of the interprofessional team in evaluating and treating patients with this condition.
Objectives:
- Summarize the etiology of incontinentia pigmenti.
- Outline the histopathology of skin lesions and the typical presentation of a patient with incontinentia pigmenti.
- Summarize the differential diagnoses of incontinentia pigmenti.
- Review the appropriate management for patients with incontinentia pigmenti.
Introduction
Incontinentia pigmenti (IP) or Bloch-Sulzberger syndrome, is a rare X-linked dominant genodermatosis. Other names of this disorder include Bloch–Siemens syndrome, Bloch–Sulzberger disease, melanoblastosis cutis, pigmented dermatosis Siemens-Bloch type, and nevus pigmentosus systematicus. IP is the consequence of a mutation in the IKBKG gene (formerly known as NEMO or nuclear factor kappa essential modulator).[1]
Incontinentia pigmenti clinically presents with skin, central nervous system, eyes, teeth, hair, and nail involvement. This rare condition usually presents within the first few weeks of life and is most commonly seen in females and rarely in males. IP is usually fatal in male infants; females show variable phenotypic presentation and survive due to lyonization, as seen in many X-linked disorders.[2] Skin involvement is one of the first to be noted and progresses through four stages: vesicular, verrucous, hyperpigmented, and hypopigmented/atrophic stage.[3] The central nervous system (CNS) manifestations and eye manifestations can cause severe disability in these patients.[4]
The diagnosis of IP can be made clinically and confirmed with genetic testing. Management of IP requires a multidisciplinary approach, including referral to pediatric dermatology for management of blisters, prevention of secondary skin infections; dental care; screening by an ophthalmologist to reduce the risk of retinal detachment; referral to pediatric neurology for management of seizures and neurological deficits; care by a pediatrician and developmental specialists to address the developmental delay.[3]
The low incidence of this condition worldwide makes diagnosis and prompt management a challenge. Surveillance and medical management protocols have been established by the Incontinentia Pigmenti International Foundation and the National Foundation for Ectodermal Dysplasias.
Etiology
Incontinentia pigmenti is a disorder of X-linked inheritance. Most affected individuals have a de novo mutation of the IKBKG (inhibitor of kappaB kinase gamma) or IKK gamma or NF-kappa-B essential modulator (NEMO) gene on chromosome Xq28.[1] IKBKG activates NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells), which is involved in apoptosis (cell survival), cell cycle, inflammation, immunity, and other pathways.[5]
Normally, NF-kB protects against cell death; it is maintained within cells in an inactive state and becomes activated to induce an inflammatory response secondary to bacterial, viral, or stress. Derangement of IKBKG results in its inability to protect cells against apoptosis; hence more cell death occurs in response to stimuli.[6]
Due to X-linked dominant inheritance, IP is usually lethal in males during embryogenesis.[7] However, cases in males have been reported in the literature. This can be explained by somatic mosaicism, hypomorphic mutations, or the presence of an extra X chromosome, as in Klinefelter syndrome. Females inherit one X chromosome from each parent, and one X chromosome becomes inactivated through lyonization. Inactivation of a single X chromosome is not seen in all cells. Therefore, females are functionally mosaic, meaning they have two cell lines. Skin lesions develop along the Blaschko's lines.[8] These lines reflect embryonic stem cells' migration path during development and are invisible unless a pigmentary skin disorder is present.[9]
Epidemiology
Due to the X-linked inheritance pattern, women affected by incontinentia pigmenti have a 50% chance of transmitting the pathogenic version of IKBKG to offspring; male fetuses with a defective gene are usually miscarried.[7] In a systematic review, 1393 cases of IP were reported from the literature analysis from 1993 to 2012.[10][11]
The number of cases both in females and males continues to grow. The prevalence of IP is estimated to be 0.7/1,000,0000 cases worldwide.[12] The majority of the living individuals with the disease are females (92 to 97%).[13] There are around 28 new cases of IP each year globally.
Pathophysiology
Incontinentia pigmenti affects all cells of ectodermal origin, including skin, hair, nails, and teeth.[14] CNS involvement is not clearly understood, but NF-kB is present in most cells, including the central nervous system, and protects the integrity of the blood-brain barrier by protecting against cell death.[15] As seen in X-linked disorders, lyonization in affected females results in functional mosaicism and manifests as cutaneous lesions in Blaschkoid distribution.
NF-kB is a transcription factor involved in expressing multiple genes, including cytokines, chemokines, growth factors, adhesion molecules, and regulators of apoptosis.[16][17] NF-kB activation prevents apoptosis induced by the tumor necrosis factor (TNF) family of cytokines. NF-kB is mainly inactive in most cells, and various stimuli trigger activation of NF-kB. These stimuli include interleukin-1 (IL-1), TNF alpha, antigen receptors (T-cell receptor and B-cell receptor), genotoxic stress (ultraviolet radiation, gamma radiation, reactive oxygen intermediates), lipopolysaccharide (bacterial endotoxin), and double-stranded RNA (viral infection).[18]
The 'canonical' NF-kB is a heterodimer consisting of p50 and p65 (RelA).[19] NF-kB is kept inactivated in the cytosol when it is complexed with inhibitory protein IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha). Various stimuli activate the IκB kinase (IKK) after attaching to and activating the receptors. IKK phosphorylates IkB, which leads to the degradation of IkB and thereby activation of NF-kB. Activated NF-kB enters the nucleus to bind with response elements (RE) of DNA (deoxyribonucleic acid) and brings out the various changes in cellular functions.[20] The mutation of IKK results in complete disruption of the signaling pathway and virtually no NF-kB activity after stimulation of NEMO/IKK-deficient cells.
Peripheral eosinophilia is noted in patients with IP. The increased production of eosinophils in the bone marrow and increased migration of eosinophils into the circulation causes this eosinophilia. An activated eosinophil secretes granulocyte-macrophage colony-stimulating factor (GM-CSF), a cytokine that induces eosinophil differentiation and maturation in the bone marrow. Activated IKK cells have been found to induce elevated expression of GM-CSF in peripheral eosinophils via the NF-kB pathway.[21]
GM-CSF works as an autocrine factor to promote eosinophil survival, further elevating peripheral counts. IL-5, a cytokine released by Th2 (T-helper type 2) cells, stimulates the production of eosinophils in the bone marrow and their release into the peripheral blood. NF-kB pathway activation indirectly stimulates the transcription of IL-5. Currently, the mechanisms of NF-kB pathway activation and T helper cell participation in IP are not precisely known. Eotaxin is thought to contribute to tissue eosinophilia in recent studies of the pathophysiology of IP. Several eotaxin promoters possess NF-kB binding sites, including one encoding an eotaxin chemokine previously isolated from blister fluid and crusted scales of patients with IP.[22]
Most recently, immunohistochemistry staining of lesional skin from patients with IP revealed strong eotaxin expression on nearly every epidermal layer. Strong staining for eotaxin was also seen in endothelial cells, and perivascular inflammatory infiltrates. A high level of eotaxin expression coincided with areas where eosinophils accumulate.
Histopathology
A skin biopsy can be done if genetic testing is inconclusive, particularly in males with suspected incontinentia pigmenti where mosaicism may be present. Histopathologic features can differ based on the stage of skin involvement:[23]
- Stage 1 (Vesicular): Eosinophilic spongiosis with intraepidermal blister formation.
- Stage 2 (Verrucous): Hyperkeratosis, acanthosis, papillomatosis with dyskeratotic keratinocytes.
- Stage 3 (hyperpigmented): Abundant melanophages with pigment incontinence, and
- Stage 4 (hypopigmented/atrophic): Atrophic epidermis with loss of rete ridge pattern and adnexal structures; and reduction in melanocytes in the basal layer.
History and Physical
Incontinentia pigmenti is a disorder of the skin, eyes, and central nervous system (CNS) that primarily affects females and rarely males. Physical examination findings involving each organ system are listed below.[24]
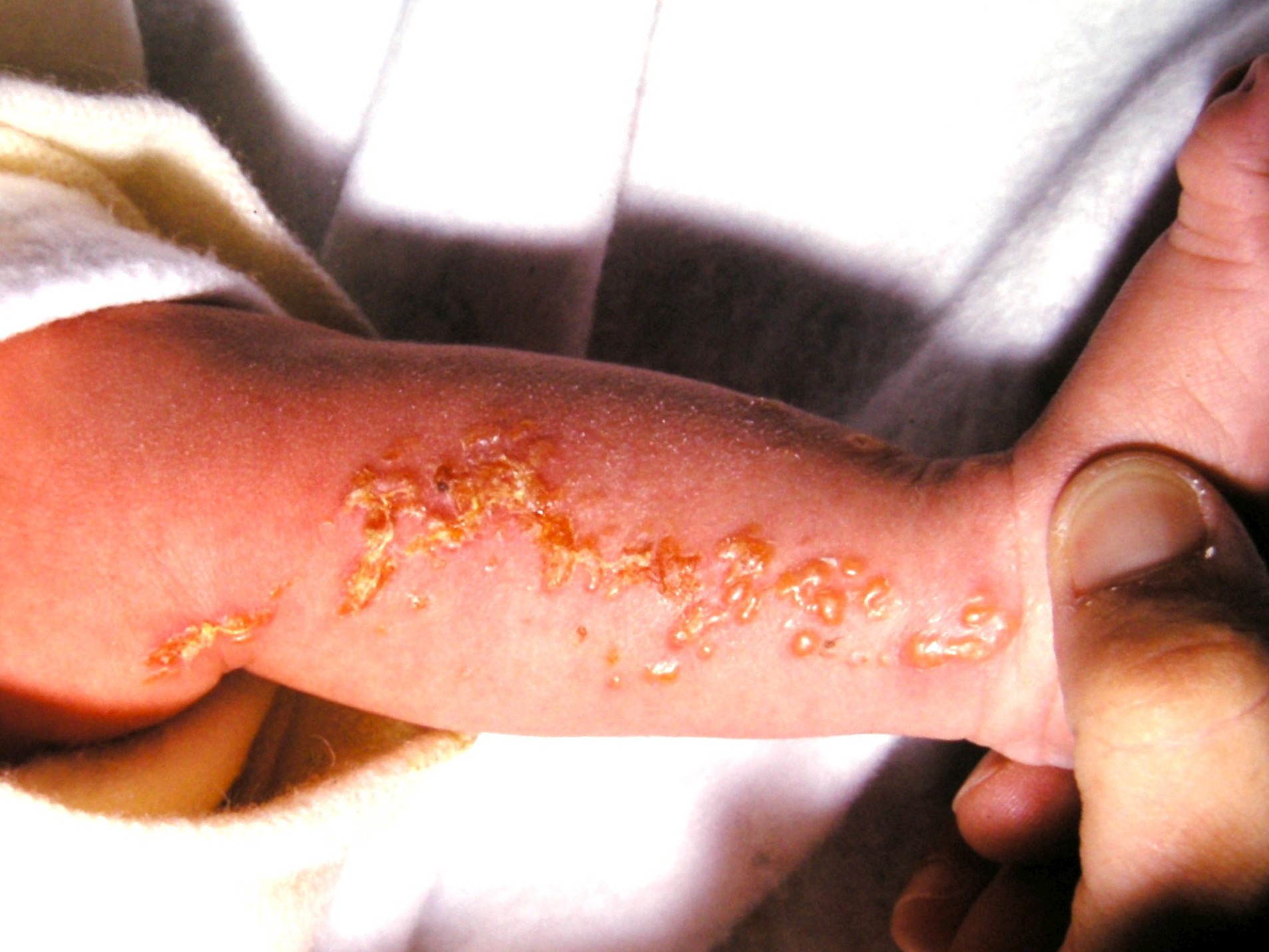
Cutaneous findings of IP are divided into four stages which overlap or occur sequentially. Dermatologic findings are the first to be observed in IP and present in nearly all patients. However, the absence of visible cutaneous lesions does not exclude the diagnosis of IP.
- Stage 1, Inflammatory Stage
- Vesicular lesions on an erythematous base following Blaschko's lines usually appear at birth or within the first eight weeks of life and persist from 2 weeks to 4 months.
- The lesions typically affect the limbs and trunk.
- These lesions usually disappear at 18 months of age. These lesions can reappear later in life, even after resolution. Reappearance is noted in the setting of febrile illnesses.[25]
- Stage 2, Verrucous Stage
- Warty papules and plaques are noted in the linear distribution along the lines of Blaschko but not necessarily along the same regions previously affected by the inflammatory stage.
- These lesions usually involve the distal limb and scalp. The skin lesions generally appear within the first few months of life as stage I lesions start to resolve.
- Stage 3, Hyperpigmented Stage
- Whorled slate grey hyperpigmented plaques (marble cake appearance) are seen following Blaschko's lines, and these slowly regress. This is the most typical appearance of IP.
- These lesions are not present at birth, and these usually occur at the age of 6 months to 1 year as stage 2 lesions start to disappear. The specific location is the trunk, axilla, or groin. However, the whole skin surface needs to be examined as the involvement may be minimal. These lesions usually disappear in adolescence.
- Stage 4, Hypopigmented Stage
- Pale, hairless linear patches and plaques devoid of sweat glands are most commonly seen on the posterior calves and arms. These lines are chronic.[26][27]
Hair: In roughly 38% of IP patients, vertex alopecia occurs most commonly in a mild manner and often goes unnoticed. Alopecia is often associated with inflammation, vesiculation, and scarring. A rare manifestation of IP is agenesis of the eyebrows and eyelashes.
Nails: Approximately 7 to 40% of patients have nail dystrophy. Ridging, pitting, or nail disruption may occur intermittently and can affect all or most of the fingernails and toenails.[28] These changes tend to regress and disappear with age. Subungual and periungual keratotic tumors usually present between puberty and the third decade. The lesions sometimes spontaneously regress but often cause pain, nail dystrophy, and, most importantly, destruction of the bone underneath.[29]
Teeth: The features include microdontia, anodontia, hypodontia, and peg-shaped teeth. Some significant dental abnormalities are microdontia, delayed eruption of permanent teeth, and cleft palate.[30]
CNS: Central nervous system deficits constitute a significant threat to the normal life span of patients with IP. When coupled with ophthalmic manifestations, these may significantly reduce patients' quality of life. Approximately 30% of IP patients have neurologic impairments. Neurologic involvement is often present along with retinal vasculopathy and may result from ischemia/vaso-occlusive events.[31]
About 13% of infants develop infantile spasms and seizure disorders. There is also an increased risk of spastic paralysis, and 7.5% of patients develop motor dysfunction. Early seizures may result from abnormal neuronal and neural crest cell migration, leading to gross brain malformations. Other manifestations include congenital hearing loss, cerebellar ataxia, muscular paralysis, mental disability, and aseptic encephalomyelitis.
Ophthalmologic: The risk of ophthalmologic abnormalities is 20 to 37%.[32][33] However, up to 77% of cases may have ocular involvement.
- Retina: Peripheral retinal capillary nonperfusion (ischemia) is noted. New vessels may give rise to vitreous hemorrhage or tractional retinal detachment. Exudative retinal detachments may also be noted. The risk of retinal detachment is most significant in infancy and childhood. The findings at the posterior pole include foveal hypoplasia, RPE (retinal pigment epithelium) mottling, areas of capillary nonperfusion surrounded by vascular loops, macular ischemia, epiretinal membrane, neovascularization of the optic disc, Roth spots, paramacular dilation of vessels, and aneurysms. The changes in the peripheral retina may also progress towards the posterior pole. The changes seen in the peripheral retina include:[34][35]
- Peripheral avascular retina
- Aneurysm like dilation
- Arteriovenous anastomosis, vascular loops similar to those seen in aggressive retinopathy of prematurity (ROP)
- Retinal and extraretinal new vessels
- Preretinal fibrovascular proliferation
- Vitreous hemorrhage
- Retinal detachment (tractional or exudative)
- Exudates
- Mottling of RPE
- Hypopigmented lesions
- Coloboma
- Other eye findings: Strabismus, cataracts, conjunctival pigmentation, optic atrophy, retinal pigment abnormalities, foveal hypoplasia, foveal disorganization, and microphthalmia are less common eye findings.[36] The end-stage disease is characterized by phthisis or absolute glaucoma. The visual decline may also be related to brain disorders leading to cortical blindness.
Breast: The presence of abnormalities of mammary tissue, ranging from aplasia of the breast, breast hypoplasia, nipple hypoplasia, to excessive nipples, may occur, and it is more common than in the general population.
Evaluation
Diagnosis of incontinentia pigmenti is based on clinical findings. Pediatricians and dermatologists are usually the first to diagnose the disease during infancy. Experts have established major and minor criteria to help make the diagnosis. Diagnostic criteria for IP have been suggested by Landy and Donnai.
If the first-degree female relative does not show evidence of IP, the major criteria include[28]
Typical Neonatal Rash
- Erythema
- Vesicles
- Eosinophilia
Typical Hyperpigmentation
- Mainly trunk
- Blaschko's lines
- Fading in adolescence
Linear, Atrophic, Hairless Lesions
Minor criteria or 'supportive evidence' in cases not showing evidence of IP in a first-degree female relative include:
- Dental involvement
- Alopecia
- Woolly hair/abnormal nails
- Retinal disease
At least one of the major criteria is necessary to confirm a diagnosis, but minor criteria support the diagnosis of sporadic IP. The absence of all minor criteria makes the diagnosis of IP 'uncertain.' If there is evidence of IP in a first-degree female relative, then the following features are needed either alone or in combination:[28]
Suggestive History or Evidence of Typical Rash
Skin manifestations of IP
- Hyperpigmentation
- Scarring
- Hairless streaks
- Alopecia at vertex
Anomalous Dentition
Woolly Hair
Retinal Disease
Multiple Male Miscarriages
Recently, the diagnostic criteria of IP have been revised to include the four stages of IP skin lesions as significant criteria. The minor criteria or the supportive evidence include 'dental anomalies, ocular anomalies, CNS anomalies, alopecia, abnormal hair (sparse hair, wooly hair, anomalies of eyebrows and eyelashes), abnormal nails, *alate anomalies, nipple and breast anomalies, multiple male miscarriages, and typical skin pathohistological findings.' According to this revised diagnostic criteria by Minic and colleagues, the conditions needed to establish the diagnosis of IP include:[10]
No evidence of IP in a first-degree female relative: If lacking genetic IKBKG mutation data, at least two or more major criteria or one major and one or more minor criteria are necessary to make a diagnosis of sporadic IP,
In the case of confirmed IKBKG mutation typical for IP: any single major or minor criterion is satisfactory for IP diagnosis,
Evidence of IP in a first-degree female relative: Any single major or at least two minor criteria,
In all cases: eosinophilia and skewed X-chromosome inactivation support diagnosis.
If clinical features are not evident, molecular genetic testing to identify IKBKG mutation is diagnostic. The most effective molecular genetic testing is single-gene testing. A skin biopsy can also be considered. Histopathologic features will vary based on the stage of clinical findings.
Ocular investigations include fundus photo, fundus fluorescein angiogram (FFA), optical coherence tomography (OCT) of the retina, and OCT-angiography (OCTA). FFA features include abnormal vascular loops, capillary nonperfusion areas, vascular anastomoses, pathologically straightened peripheral retinal vessels, and new vessels.[37] Abnormalities seen on OCT macula include irregularities in the outer plexiform layer, thinning of the inner retina, epiretinal membrane, and macular pseudohole. OCTA abnormalities are noted in almost all eyes. The abnormalities include abnormal vascular loops, loss of flow in deep and superficial capillary plexuses (corroborating with retinal thinning seen on OCT), and reduced vascular density.[38]
Neuroimaging plays an important part in the diagnosis of CNS involvement.
Treatment / Management
The involvement of multiple organ systems in incontinentia pigmenti requires a multidisciplinary approach to management.
- Pediatric dermatology should be consulted for the management of cutaneous lesions.
- An early blistering rash can be misdiagnosed as congenital herpes simplex, epidermolysis bullosa, and varicella.
- All stages of skin lesions should be kept cool and dry, and devoid of trauma.
- All stages of skin lesions can overlap in the first year of life and recur after the disappearance in the setting of infection or viral illness.
- Hypohidrosis occurs in IP, which results in heat intolerance and the risk of life-threatening overheating.[25] Therefore, measures for cooling should be undertaken, including cool baths, use of ice packs, and shade.
- No specific therapies have been established for cutaneous lesions. Treatment is focused on wound care and treatment of secondary infections as needed.
- A pediatric ophthalmologist should be consulted to examine the retina and periodic follow-ups.
- A dilated fundus examination should be performed to evaluate the optic nerve, macula, and retinal periphery.
- A retina specialist should re-evaluate abnormal findings
- The goal is to prevent retinal detachment or vitreous hemorrhage secondary to retinal neovascularization.
- The peripheral avascular retina is treated with laser photocoagulation similar to retinopathy of prematurity.
- Retinal detachment and vitreous hemorrhage may require pars plana vitrectomy.
- Pediatric neurology consultation is needed for the management of seizures and neurologic imaging.
- Baseline magnetic resonance imaging (MRI) with or without contrast during the neonatal period is recommended to establish baseline neurologic imaging.
- Repeat imaging in seizures and other neurologic abnormalities can be performed.
- Seizures are the most common neurologic manifestation. An electroencephalogram (EEG) may be considered in the setting of seizures. A periodic follow-up to assess for focal motor deficits and delay in developmental milestones is necessary.[15]
- A genetic counselor and a geneticist can help in genetic testing. Approximately 25 to 35% of cases are familial, and the rest are sporadic. A thorough exam of patients and at-risk relatives can help determine inheritance. If there are no physical findings, molecular genetic testing for a mutation in the IKBKG gene can be done if the diagnosis is suspected.
- Follow-up with the pediatrician is necessary to screen for developmental delays. Neuropsychiatry testing for children who are struggling with school activities is essential.
- Other systemic complications include damage to the microvasculature and pulmonary hypertension without cardiovascular disease.
- In pregnancy, fertility is not impaired in women with IP, but the risk of spontaneous abortion is higher for those carrying a male fetus.
- Women with IP considering pregnancy should have prenatal testing and counseling. Males with IP have somatic mosaicism, not a germline mutation; therefore, transmission from an affected male to a daughter is unlikely.[3]
Differential Diagnosis
Differential diagnoses of incontinentia pigmenti can vary based on the stage of skin lesions. These include:[39]
Stage 1 (vesicular)
- Congenital herpes simplex can present with grouped hemorrhagic vesicles, often on the scalp and at sites of trauma from scalp electrodes. Lesions can be present at birth or appear within the first few weeks of life.[40]
- Bullous impetigo,
- Epidermolysis bullosa, and
- Congenital ichthyosiform erythroderma.
Stage 2 (verrucous) - linear epidermal nevus and lichen striatus
Stage 3 (hyperpigmented) - linear and whorled nevoid hype melanosis
Stage 4 (atrophic/hypopigmented) - hypomelanosis of Ito, Goltz syndrome( focal dermal hypoplasia).
Differential diagnoses of retinal features of IP include ROP, FEVR (familial exudative vitreoretinopathy), and Norrie disease. IP is an important cause of leukocoria.[41]
Staging
Vitreoretinal changes noted in IP are classified into five stages:[34]
Stage 1- Retinal pigment epithelial changes only,
Stage 2 - Retinal vascular abnormalities (except neovascularization),
Stage 3 - Retinopathy with neovascularization or other pathologies secondary to retinal vasculopathy, such as retinal exudation, epiretinal membrane/proliferation, or vitreous hemorrhage,
Stage 4 - Retinal detachment without end-stage changes
- 4a: partial retinal detachment;
- 4b: total retinal detachment, and
Stage 5 - End stage: severe ocular complications such as phthisis bulbi and secondary glaucoma.
Prognosis
Prognosis is variable and based on the degree of skin and organ involvement. There are no significant sequelae secondary to skin manifestations. However, the severity of eye and CNS abnormalities pose the highest morbidity and mortality. In addition, patients with seizures and structural CNS abnormalities have the greatest developmental delay and impairment risk. Patients without significant ophthalmic or neurological impairment have a good prognosis and a normal life expectancy.[42]
Complications
In a newborn, the goal is to reduce the risk of infection of blisters, in addition to standard medical management of skin lesions. Careful monitoring for systemic involvement is prudent during this time. The possibility of retinal detachment, particularly in children under seven years of age, is an important consideration, and ophthalmologic screening is necessary. Head trauma can also increase the risk of retinal detachment; however, no specific recommendations are currently established for avoiding contact sports. A suggested schedule for eye examinations is monthly until age four months, every three months from 4 months to 1 year of age, then every six months between age one to three, and annually after that. The neurologic function requires routine assessment, and ongoing dental screening is appropriate.[4]
Deterrence and Patient Education
Support and advocacy groups help patients and families find valuable services. To establish the best professional and social resources, it is imperative to seek help from experts and receive an early diagnosis. Rare diseases are challenging to diagnose and require genetic testing and a multidisciplinary approach. For families with limited health literacy or resources, this may be a challenging undertaking. Dermatologists and pediatricians are uniquely positioned to help patients find the right support network as they are often a patient's first point of contact.
Patients and families can learn more about IP by visiting the following websites of the following organizations:
- Incontinentia Pigmenti International Foundation
- National Organization for Rare Diseases
Enhancing Healthcare Team Outcomes
Providing medical care and support to individuals and families with incontinentia pigmenti requires a multidisciplinary approach. Follow-up with multiple specialties includes ophthalmology for annular eye exams throughout life and geneticist for risk assessment for older and younger at-risk relatives. Rare diseases like IP have an overall low incidence, so establishing treatment protocols is difficult. Support for families can also prove to be a challenge due to this.