Introduction
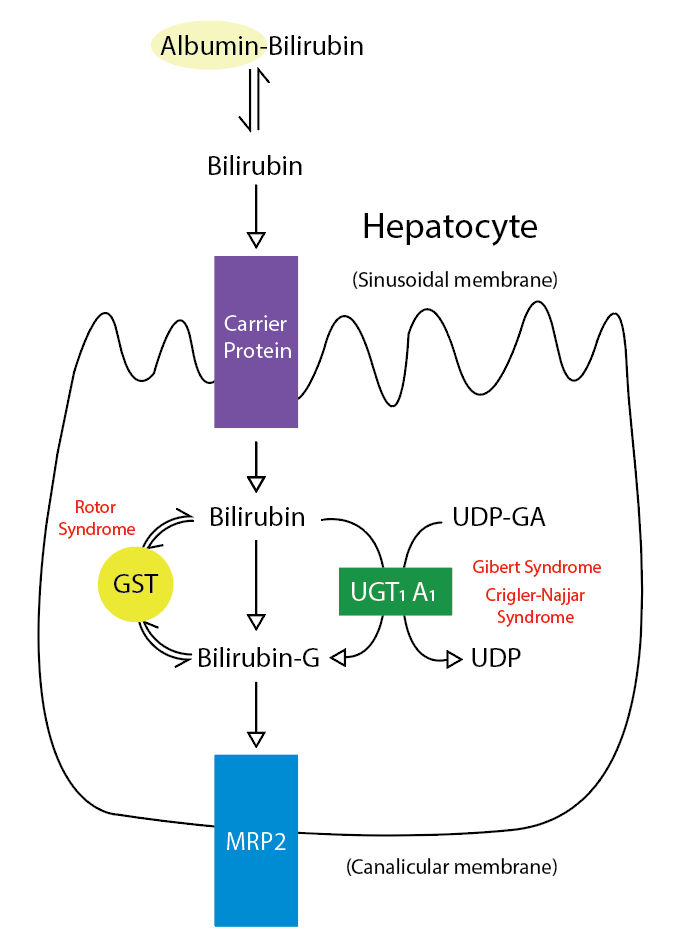
Bile is a fundamental and unique secretion of the liver. It is a yellow-greenish fluid secreted from the hepatocytes and altered as it passes through the biliary tree by the epithelial cells lining the bile duct (cholangiocytes). After its secretion, bile is stored and concentrated in the gallbladder. Lipids in the duodenum stimulate the release of cholecystokinin, which stimulates gallbladder wall contraction, resulting in the release of bile into the cystic and common bile duct.[1] The bile is composed mainly of water electrolytes and other substances, which include bile salts, cholesterol, bilirubin, lecithin, amino acids, drugs, toxins, heavy metals, and vitamins (see Image. Metabolic Pathway for Bilirubin in the Hepatocyte).[1] Finding some hormones, proteins, and peptides in the bile secretions is also possible.[2]
Cellular Level
The Golgi apparatus, microtubules, and microfilaments in the hepatocytes participate in bile formation. The Golgi complex provides the vesicles for the substances secreted into the bile. The ATP-binding-cassette (ABC) that is newly synthesized is transferred from the Golgi complex to the canalicular membrane to function as a transporter. This process needs intact microtubules and microfilaments.[2]
Functionally the liver might be divided into 3 zones. Zone 3 is the zone that is around the central vein, while zone 1 is the zone that is most distal from the central vein and surrounds the portal triad (portal vein, hepatic artery, and bile duct); hence, it is also called the periportal zone. Zone 2 lies between zone 3 and zone 1.[2]
Estimates are that the bile flow in humans averages 620 mL/day. Bile flow is either dependent or independent of the osmotic force of bile acids. When the bile flow is dependent on the osmotic force of bile acids, it is the bile-acid dependent fraction of bile (BADF), while if the flow is not dependent on the osmotic force of bile acids, it is bile acid-independent fraction of bile (BAIF). Moreover, there is a linear relationship between the amount of water that follows bile acids osmotically and the bile acids in the bile. Certain bile acids retain the capacity to induce a higher volume of bile (hypercholeresis). One classic example is ursodeoxycholic acid. There is a proposal that the hyperocholeresis might be due to the reabsorption of ursodeoxycholic acids from the epithelial cells that line the bile ducts into the bloodstream and then into the hepatic sinusoids. The hepatocytes take these hypercholeretic bile acids from the hepatic sinusoids. In the hepatocytes, the hypercholeretic bile acids are re-secreted into the bile canaliculi, thereby increasing the bile-acid-dependent fraction of bile flow and resulting in the so-called hypercholeresis. This pathway is called the cholehepatic shunt pathway. The bile acids in the BADF are either newly synthesized in the liver or else they come from the enterohepatic circulation of bile acid.[2] Of note, ursodeoxycholic acid (hypercholeretic bile acid) is used in primary biliary cholangitis (formerly cirrhosis), a condition with cholestasis and decreased bile secretion.
The mediation of the uptake of bile acids from the sinusoids is via Na-dependent bile acid transporters and Na-independent bile acid transporters. The Na-dependent bile acid transporter is called sodium taurocholate-cotransporting polypeptide (NTCP). NCTP does not express at the canalicular membrane; it only expresses at the basolateral membrane. The Na-independent mechanism involves organic anion transporting polypeptides (OATPs) transporter. OATP transporter transports bile acids into the hepatocytes in exchange for bicarbonate or glutathione. Certain Na-independent bile acid transporters (OST/alpha and OST/beta) are present at the basal membrane of cholangiocytes and hepatocytes. In the cholangiocytes, these transporters may participate in the cholehepatic shunt pathway (explained above), while in the hepatocytes, these transporters may participate in bile acid efflux into the bloodstream in cases of bile accumulation in the hepatocytes (cholestasis). After the uptake of bile acids from the sinusoids or after they are synthesized endogenously from cholesterol, the unconjugated and conjugated bile acids get secreted into the canaliculi through a special energy-dependent transporter called bile salt export pump (BSEP). BSEP not only transports bile acids into the canaliculi, but it also aids in the excretion of certain drugs. The BAIF of bile flow is mainly determined by the ATP-dependent transport of glutathione through the multidrug resistance-associated protein 2 (MRP2). Cholesterol and sterols are secreted by ABCG5/G8 protein, while phospholipids and lipophilic cationic drugs are secreted by multidrug resistance-P glycoprotein 3 (MDR-3) and MDR1, respectively.[2]
Hormonal and neural mechanisms regulate the secretion of bile. The hormonal mechanism involves secretin, somatostatin, dopamine, and gastrin, while the neuronal mechanism involves acetylcholine through the vagus nerve. A vasoactive intestinal polypeptide (VIP), in part, also increases secretin-mediated bile flow. Secretin increases the secretion of sodium bicarbonate-rich fluid by binding to its receptors at the basolateral side cholangiocytes. Dopamine, gastrin, and somatostatin inhibit secretin-mediated sodium bicarbonate-rich fluid secretion.[2] The secretion of an alkaline fluid is the function of the cholangiocytes.
Development
The liver tissue develops as a diverticulum from the ventral foregut endoderm. The ventral foregut endoderm has 2 portions, the cranial and caudal portions. The intrahepatic duct develops from the cranial portion, while the extrahepatic bile ducts develop from the caudal portion of the ventral foregut endoderm.[3][4][5]
Around the eighth week of gestation, the epithelium of the intrahepatic duct begins to develop, and centrifugally, it proceeds from the hilum to the periphery of the liver. The intrahepatic biliary epithelium becomes completely mature only during the first year of life.[6][7][8]
The extrahepatic biliary epithelium develops before the intrahepatic duct. Both of the ducts combine at the level of the hepatic hilum.[9]
Organ Systems Involved
The organ systems involved are the hematologic, gastrointestinal (GI), and hepatobiliary systems. The hematologic system contributes through the formation of bilirubin from biliverdin. Biliverdin forms from the oxidation of heme in old RBCs in the reticuloendothelial system. The GI system is responsible for regulating the alkaline secretions of bile, secreting CCK which causes contraction of the gallbladder, active absorption of bile salts in the terminal ileum, excretion of cholesterol, and excretion of bilirubin in the form of stercobilin. Stercobilin forms by the action of the colonic bacterial flora, which converts urobilinogen into stercobilin. The hepatobiliary system is responsible for bile formation, secretion, and storage.
Function
The bile has 5 main functions:
- Aids in the digestion of fat via fat emulsification
- Absorption of fat and fat-soluble vitamins
- Excretion of bilirubin and excess cholesterol
- Provides an alkaline fluid in the duodenum to neutralize the acidic pH of the chyme that comes from the stomach
- It provides bactericidal activity against microorganisms present in the ingested food
Mechanism
Bile is synthesized in the hepatocytes and secreted into the bile canaliculi until it reaches the sphincter of Oddi. If the sphincter closes, the bile is refluxed back into the gallbladder to be stored and concentrated there. Bile becomes concentrated by the reabsorption of water and electrolytes and, to some extent, by the reabsorption of bile salts. Once the food is present in the duodenum (especially fatty food), the I cells are stimulated to secrete CCK which in turn causes gallbladder wall contraction as well as relaxation of the sphincter of Oddi. The bile then flows into the second part of the duodenum and causes the emulsification of large fat droplets into small ones. The formation of micelles aid in the absorption of fat and fat-soluble particles.; this is because the micelle has a central hydrophobic core and peripheral hydrophilic form. The fat and fat-soluble vitamins become incorporated into the central hydrophobic part of the micelle for reabsorption.
Related Testing
Testing for bile pathology includes imaging and blood tests. Imaging tests include ultrasonography for gallbladder pathology (eg, cholecystitis and gallstones), Magnetic resonance cholangiopancreatography (MRCP) for visualization of the biliary tree (eg, primary sclerosing cholangitis), abdominal computed tomography (CT), abdominal magnetic resonance imaging (MRI), and hepatobiliary iminodiacetic acid (HIDA) scan. HIDA scan is particularly useful in confirming acute cholecystitis when ultrasonography is inconclusive. The absence of gallbladder visualization on the HIDA scan suggests acute cholecystitis. Blood tests include CBC and liver function tests (LFT).
CBC may show leukocytosis accompanying many hepatobiliary pathologies (eg, cholecystitis and ascending cholangitis). LFT includes alkaline phosphatase (ALP) and gamma-glutamyltransferase (GGT), aspartate aminotransferase (AST) and alanine aminotransferase (ALT), prothrombin time (INR), indirect (unconjugated bilirubin), direct (conjugated) bilirubin, total bilirubin, and serum albumin.
Marked elevation in GGT and ALP with direct hyperbilirubinemia with or without a minimal elevation in AST and ALT indicates a cholestatic pathology. Normal LFT, except for high ALP, would point to a bone pathology rather than a hepatobiliary pathology. Markedly elevated GGT with slightly abnormal LFT would point to an infiltrative liver pathology (eg, metastasis).
Pathophysiology
The biliary system pathology includes autoimmune diseases, obstructive cholestasis, non-obstructive cholestasis, infections, and malignancy. The autoimmune diseases are primary biliary cholangitis (PBC) and primary sclerosing cholangitis. There is autoimmune-mediated progressive destruction of intrahepatic small-medium-sized bile ducts in primary biliary cholangitis, resulting in impaired bile secretion and intrahepatic cholestasis.[10] There is intrahepatic and extrahepatic destruction of bile ducts in primary sclerosing cholangitis. PSC is strongly associated with ulcerative colitis. The autoimmune basis for both conditions (PBC and PSC) is present in the histologic findings and specific antibodies in patients with these 2 conditions. Moreover, both conditions are often associated with other autoimmune diseases, further supporting the autoimmune basis.
Cholestasis occurs whenever there is impaired bile secretion from the hepatocytes or obstruction to the bile flow. The obstruction might be due to gallstones (most common) or any other cause, such as external compression of the biliary tree or narrowing of the biliary tree, as seen in PSC. Adenocarcinoma of the head of the pancreas is an important cause of painless jaundice secondary to obstruction of the bile duct by the enlarging head of the pancreas. Once cholestasis occurs, bilirubin accumulates in the body resulting in yellowish discoloration of the sclera and skin. Moreover, the accumulation of bile acids secondary to cholestasis might be responsible for the pruritus seen in these patients.
Infections of the bile ducts are not uncommon, and they might present as fever, right upper quadrant pain, and jaundice. Inflammation of the gallbladder (cholecystitis) is also common and usually associated with gallstones (ie, acute calculous cholecystitis).
Clinical Significance
Understanding the pathophysiology of jaundice and how bile is synthesized and secreted is essential to making a proper diagnosis and providing the proper treatment. Signs and Symptoms of biliary tract pathology include jaundice, pruritus, fever, xanthomas, steatorrhea (due to fat malabsorption), right upper quadrant pain (RUQ), and fatty dyspepsia (a feature of bile obstruction) (10). Biliary tract pathology may decrease the absorption of fat and fat-soluble vitamins (A, D, E, and K). A deficiency in vitamin K would cause bleeding, while vitamin D deficiency may cause rickets in children and osteomalacia in adults due to decreased calcium absorption from the gut. Deficiency in vitamin A may lead to night blindness.