Continuing Education Activity
Lymphoma of the central nervous system, both primary and secondary, represents a rare subset of non-Hodgkin lymphoma. Primary central nervous system lymphoma refers to those cases confined to the CNS parenchyma, dura, leptomeninges, cranial nerves, and spinal cord or the intraocular compartment in immunocompetent patients. On the other hand, secondary CNS lymphoma refers to systemic non-Hodgkin's lymphoma that has disseminated to the CNS. Historically, the prognosis of primary central nervous system lymphoma has been very dismal, with overall survival of 1.5 months when untreated, and a 5-year survival rate of 30%. This activity describes the pathophysiology of CNS lymphoma and highlights the role of the interprofessional team in its management.
Objectives:
- Identify the epidemiology of central nervous system lymphoma.
- Review the evaluation of central nervous system lymphoma.
- Outline the treatment and management options available for central nervous system lymphoma.
- Describe interprofessional team strategies for improving care coordination and communication to advance central nervous system lymphoma and improve outcomes.
Introduction
Lymphoma of the central nervous system (CNS), both primary and secondary, represents a rare but highly aggressive subset of non-Hodgkin lymphoma.[1]
CNS lymphoma consists of 2 major subtypes:
- Primary central nervous system lymphoma (PCNSL), and
- Secondary CNS involvement by systemic lymphoma.[2]
PCNSL is a rare variant of extra-nodal non-Hodgkin lymphoma (NHL), which involves the neuraxis including the orbit, brain, leptomeninges, and spinal cord.[1] On the other hand, secondary CNS lymphoma refers to systemic non-Hodgkin lymphoma that has disseminated to the CNS. Historically, the prognosis of primary central nervous system lymphoma has been very dismal, with overall survival of 1.5 months when untreated, and a 5-year survival rate of 30%.[3][4] Meanwhile, patients with aggressive systemic non-Hodgkin’s lymphoma have a 2 to 27% risk of developing secondary CNS dissemination, with a median survival of 2.2 months after diagnosis.[5] Due to the introduction of high-dose methotrexate-based chemotherapy regimens, there has been substantial progress in treating patients with lymphomas of the CNS, leading to improved survival.
Etiology
Immunodeficiency, both primary and acquired, is a significant risk factor for lymphoma of the central nervous system.[1][5][6] Primary CNS lymphoma is seen in approximately 6% of patients with AIDS and is an AIDS-defining illness.[7] It usually occurs when CD4 counts are very low, often in patients not on antiretroviral therapy. Meanwhile, 2 to 7% of cardiac, liver, and lung transplant patients, as well as up to 2% of renal transplant patients, ultimately develop the disease. The incidence is highest during the first year post-heart and lung transplant.[6] Primary immunodeficiencies such as Wiskott Aldrich, ataxia telangiectasia, common variable, and severe combined immunodeficiency syndromes confer a 4% risk of developing primary central nervous system lymphoma.[5] The Epstein bar virus (EBV) highly correlates with CNS lymphoma in T cell immunodeficient states, such as patients on immunosuppressants post-organ transplant. EBV is also associated with 100% of primary CNS lymphoma in patients with AIDS.[5]
Epidemiology
Primary CNS lymphoma has an annual incidence of approximately 1500 cases in the United States.[1][8] It comprises 3% of all primary brain tumors and 1% of all cases of non-Hodgkin lymphoma with an incidence rate of approximately 0.5 per 100,000 per year.[1][7] While CNS lymphoma is decreasing in the AIDS population due to the advent of highly active antiretroviral therapy (HAART), its incidence is rising in the elderly population. Rates are highest among the elderly and individuals with a compromised immune system, but the disease is rare in pediatric populations. Immunocompetent patients are often diagnosed between ages 50 to 70, while immunocompromised patients present earlier in their 30s and 40s.[6] Males are more frequently affected than females in both groups, but there is no gender predilection of CNS lymphoma in patients with post-transplant lymphoproliferative CNS lymphoma.[6][9] The median age at diagnosis is approximately 65 years.[7]
Pathophysiology
The exact pathogenesis of their neurotropism is debatable.[4] The CNS that lacks any lymph nodes or lymphatics paradoxically does develop B cell non-Hodgkin’s lymphomas by following postulated mechanisms:
- Acquiring new surface markers to “home” inside the CNS, and
- Getting transformed during their intravascular transit to the CNS.
Systemic lymphoma spreads to the CNS via hematogenous dissemination.
The most commonly involved sites of primary central nervous system lymphoma are the frontal lobe and basal ganglia, with the brainstem, cerebellum, and spinal cord less commonly affected. Up to 25% of patients with primary central nervous system lymphoma develop intraocular lymphoma, and primary intraocular lymphoma ultimately disseminates to the CNS more than 80% of the time. Concurrent involvement of cerebrospinal fluid (CSF) and orbit occur in up to 20% of cases respectively.[7] Lesions of primary intraocular lymphoma tend to be found more often within the vitreous fluid and the retina.[9] However, it is uncommon for primary CNS lymphoma to disseminate systemically. On the other hand, secondary lymphoma of the CNS often has a predilection for the dura and leptomeninges, and the choroid in cases of intraocular involvement. The metastatic form that involves the leptomeninges is observed in 4-11% of patients with systemic lymphoma and 40% of lymphoma occurs in the vicinity of the CNS, such as orbital or paranasal sinuses. They seldom form nodules (non-cohesive) and cause multifocal involvement along the neuraxis leading to predominant cranial neuropathies and radiculopathy. Epidural metastasis is seen in 0.1%-6.5% of cases.
The hallmark gene “signatures” of PCNSL comprise:
- Germinal center B-cell
- Activated B-cell, and
- Type 3 large B-cell.[4]
They mirror the post-germinal center or an activated B-cell (ABC) immuno-phenotype (CD10–, BCL-6+, MUM1/IRF4+).[7] They characteristically bear pan B-cell antigens (CD19, CD20, and CD79a).[7]
Several mutations in tumor suppressor and proto-oncogenes involved in B cell activation, differentiation, and apoptosis are believed to contribute to the development of primary CNS lymphoma. Somatic hypermutations in proto-oncogenes such as MYC, PAX5, Rho/TTF, and PIM1 as well as the tumor suppressor genes such as PRDM1 have been demonstrated in primary CNS lymphoma cases. NF-KB signaling is believed to play a role in disease pathogenesis. The up-regulation of activators within the NF-KB pathway such as MYD88, CADR11, and CD79 as well as suppression of NF-KB inhibitors such as TNFAIP3 have presented in individuals with primary CNS lymphoma.[4] Expression of MYD88 and CD79B mutations in secondary CNS lymphoma is considerably lower than in primary CNS lymphoma, which suggests different pathogenesis.[10] Aberrant regulation of the JAK/STAT pathway is also implicated. Increased levels of IL-10, A JAK/STAT mediator are seen in CSF analysis of primary CNS lymphoma patients and are often associated with a worse prognosis. It is still unclear whether cells of primary CNS lymphoma first arise from within the CNS itself, or rather systemically then travel to the CNS via peripheral blood and further accumulate other mutations.[4]
The salient phenotypical alterations occurring in CNS lymphomas can be summarized as:
- Interleukin-4 (IL-4) signaling pathways with mediators such as X-box binding protein 1 (XBP-1) and signal transducer and activator of transcription 6 (STAT6)
- NFκB mutations in MYD88 and CD79B.
- Programmed death ligand 1/programmed death ligand 2 locus suggests the role of immune evasion.
- Alterations in copy number and translocations in 9p24.1.
- Up-regulation of the programmed cell death 1 receptor (PD-1) ligand.
- Loss of human leukocyte (HLA) class I or II surface antigens.
- Differential expression of MUM1.[4][7]
Histopathology
About 95% of primary CNS lymphomas belong to the diffuse large B cell (DLBC) category, with low-grade B cell lymphoma, T cell lymphoma, and Burkitt lymphoma accounting for the rest.[4][7][11][12] There are two histological variants of diffuse large B cell lymphoma: the germinal center subtype, often CD10 and BCL6 positive, and the activated B cell subtype which usually expresses MUM1. More than half of primary CNS lymphomas express both BCL6 and MUM1, suggesting that the tumor derives from B cells in the process of exiting the germinal center but that has not yet reached the post-germinal center stage. While primary CNS lymphomas usually express the pan B cell markers CD19, CD20, CD22, and CD79a, the plasma cell markers CD38 and CD138 are often absent. Only 10% of cases are CD10 positive, but 80-90% of tumors are MUM1 positive, and 60-80% are BCL6 positive. While the prognostic value of these B cell differentiation markers is well elucidated in systemic diffuse large B cell lymphomas, their significance in primary CNS lymphoma remains unclear. Similarly, overexpression of the cell cycle regulator protein MYC, as well as the anti-apoptosis protein BCL2, has prognostic implications in systemic DLBC lymphoma but is of uncertain significance in primary CNS lymphomas.[4]
Primary CNS lymphoma is a highly cellular and infiltrative tumor. Histology often displays a perivascular growth pattern called angiotropism (angiocentric patterns of lymphoid clustering).[12][13] The lymphomatosis cerebri variant of diffuse large B cell lymphoma is rarely seen. A reactive perivascular infiltrate of T cells when present is associated with a better outcome. This antitumor T cell immune response is seen less in primary CNS lymphomas when compared to systemic DLBC lymphomas, which could in part account for its poorer prognosis.[4]
History and Physical
Up to 80% of patients with primary CNS lymphoma present with focal neurologic deficits and symptoms often correlate with the location of the lesion.[5][9] Cognitive impairment is the most frequent clinical presentation, followed by gait disturbances, focal neurologic deficits, symptoms of increased intracranial pressure, and seizures.[7]
They present as focal mass lesions in almost 50% of cases and present predominantly with:
- Neuropsychiatric signs
- Features of raised intracranial pressure
- Seizures
- Ocular symptoms (pain, decreased visual acuity, photophobia, blurry vision, floaters), and
- Rarely with B symptoms.[12][13]
The predominant anatomical locations of involvement include:
- Cerebral hemisphere
- Thalamus/basal ganglia
- Corpus callosum
- The periventricular region, and
- Cerebellum.[12]
Neurologic signs and symptoms develop acutely with the following patterns of cranial and extra-cranial involvements:
- Frontoparietal lobes (39%)
- Eyes (15% to 25%)
- CSF (7% to 42%) and
- Rarely the spinal cord.[13]
Evaluation
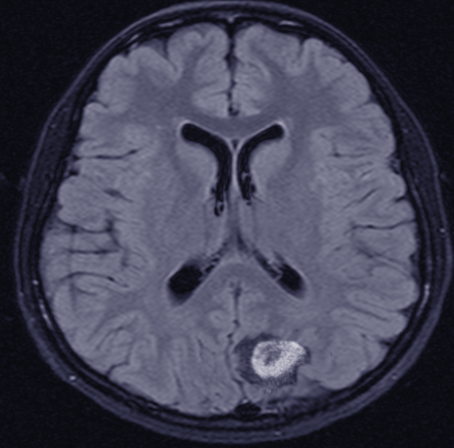
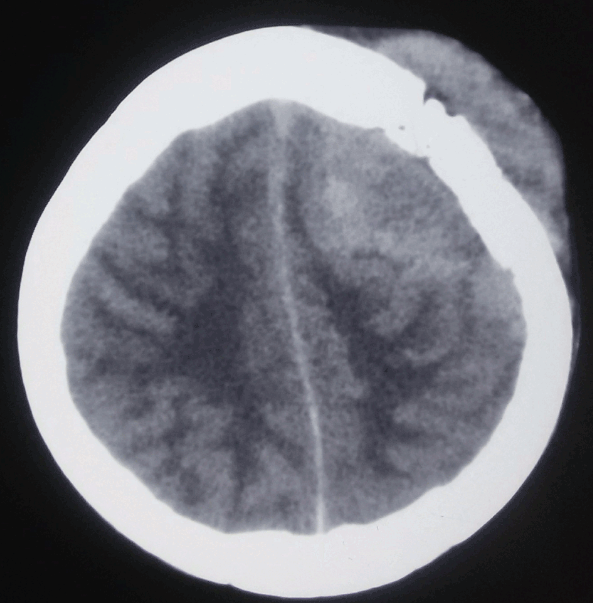
Most immunocompetent patients have solitary brain mass, with multiple lesions observed in 20–40% of cases.[7] In patients with suspected primary central nervous system lymphoma, an MRI brain with contrast is the recommended first test in diagnosis. Lesions are often located centrally within the cerebral white matter as well as in the periventricular region. They are often isointense or hypointense on unenhanced T1 weighted imaging and isointense or hyperintense on T2 sequences.[7] These lesions typically show homogeneous enhancement (contrary to HIV-associated PCNSL which are ring-enhancing).[14] Conventional magnetic resonance imaging alone cannot reliably differentiate CNS lymphoma from other neoplastic lesions in the brain, so other sequences and imaging modalities are often helpful. Primary central nervous system lymphomas are highly cellular tumors, which makes them appear restricted on diffusion-weighted (DWI) MRI sequences when compared to metastases and high-grade gliomas. Corresponding apparent diffusion coefficient (ADC) values are also lower and may be predictive of overall survival. However, AIDs-related CNS lymphomas have similarly low ADC values when compared to cerebral toxoplasmosis and cannot be reliably differentiated based solely on ADC values. In this case, MR spectroscopy may offer some benefits as lymphoma and toxoplasmosis have different biochemical properties. MR spectroscopy (MRS) shows increased choline, lipid, and lactate peaks, whereas decreased N-acetyl aspartate (NAA), and creatine peaks.[7]
MRI has a low sensitivity for detecting intraocular lymphoma, so a thin section protocol is required to reveal any nodular enhancing lesions on the macula or uvea.[14] While the preferred method for definitive diagnosis of primary vitreoretinal lymphoma is a vitreous aspiration or chorioretinal biopsy, a slit-lamp examination of both eyes is often done earlier in the workup process.[3][9][14] Flow cytometry can be used in the analysis of vitreous aspirate as well as in CSF analysis in cases of primary dural lymphoma, posterior fossa lesions, or secondary CNS lymphoma.[1][5] CSF and vitreous specimens should be assessed by flow cytometry, cytology, and immunoglobulin heavy-chain gene rearrangement.[7]A clonal IGH rearrangement in a polymerase chain reaction has a diagnostic implication.[1] Lumbar puncture for CSF cytomorphology and flow cytometric analysis has a diagnostic yield of 6–13.3%.[7]
CT is not as sensitive as MRI but usually shows iso or hyperattenuating lesions owing to hyper-cellularity and high nucleus-to-cytoplasm ratio.[7] While lesions in immunocompetent patients are usually solitary with homogenous enhancement, 20% to 40% of cases report multiple lesions, and ring-like enhancement occurs in up to 13% of cases. Surrounding edema is often present but not to the extent seen in malignant gliomas or metastatic disease. Additionally, linear enhancement along perivascular space is highly associated with primary central nervous system lymphoma. In contrast, 30% to 80% of immunodeficient patients present with multiple lesions that usually have necrosis resulting in an irregular ring-enhancing pattern post-contrast.[5][9] In the primary dural subtype of primary CNS lymphoma, diffusely enhancing masses are usually visible on CT and MRI that can mimic a meningioma. While micro-hemorrhages often present in high-grade gliomas, hemorrhage and calcifications are uncommon in primary CNS lymphoma lesions except in AIDs-related cases.
Additionally, CNS lymphomas are usually more hypermetabolic than gliomas, resulting in increased uptake on metabolic imaging such as Positron emission tomography (PET) as well as fluorodeoxyglucose (FDG) PET scans. The uptake area on PET imaging is often larger than the corresponding areas on conventional imaging, which reflects the infiltration of the tumor beyond the areas depicted on MR imaging. On the other hand, infectious lesions are generally hypometabolic, corresponding to lower thallium-201 uptake on SPECT and SPET as well as lower FDG uptake on PET imaging. This aids in the differentiation of primary central nervous system lymphomas in immunocompromised individuals from infectious etiologies. In patients without systemic involvement in contrast-enhanced CT, F-Fluorodeoxyglucose-PET revealed systemic PCNSL in 8%.[7]
The salient radiological characteristics of these lesions include:
- High cellularity which is observed as restricted in DWI and decreased ADC)
- Blood-brain barrier disruption in Dynamic susceptibility and contrast-enhanced images (DSC)
- High glycolytic metabolism in PET scan, and
- Absent angiogenesis is observed as normal cerebral blood volume (CBV) in DSC.[15]
The stereotactic biopsy is the mainstay of diagnosis.[1] Stereotactic or frameless biopsy has a diagnostic yield of more than 91%- can be aided by frozen section and 5-ALA fluorescence.[7]
When a biopsy is not possible, a diagnosis can be supported by the:
- MRI findings
- Clinical features
- Clonal B cells expansion in CSF or vitreous fluid flow cytometry, and
- IGHV gene rearrangements in PCR.[1]
Radiomics and liquid biopsy are emerging new modalities for diagnosis.[1][7][15] CSF analysis for CD79B and MYD88 or diagnostic markers like CXCL-13, B2M, and neopterin has shown promising results.[7] miR-21, sIL-2R in blood; and CXCL-13, B2M and neopterin in CSF are new markers.[2]
The International PCNSL Collaborative Group has advocated the following recommendations during diagnostic evaluation for PCNSL:
- Lymph nodes as well as testicular examination
- Contrast-enhanced MRI or computed tomography brain of the complete neuraxis
- Lumbar puncture
- Ophthalmologic examination
- Contrast CT of the chest, abdomen, and pelvis
- Bone marrow biopsy, and
- Testicular ultrasonography in older age groups.[1][7]
Treatment / Management
The dictum of management in CNS lymphoma is:
- Prolonging remission
- Reducing refractory cases
- Increasing management strategies for recurrent PCNSL.[13]
The management strategies should take into consideration:
- Physiological fitness
- Performance status
- Neurocognitive function
- Previous therapy and patterns of response, and
- Patient choice.[1]
Management strategies during remission and consolidation phases include:
- Corticosteroids
- Chemotherapy, and
- Whole brain Radiotherapy (WBRT).[1][7]
The mainstay of management and the most time-critical step is to reduce the diagnostic delay.[7]
Complete surgical resection is not amenable due to its infiltrative nature, multifocality, and microscopic seedlings protected by the intact blood-brain barrier.[1] There is no conclusive current evidence for advocating aggressive surgical removal of these lesions.[1] No clear evidence of gross resection offering any outcome advantages.[16] Extensive resection of PCNSL lesions is only advocated during features of herniation.[1]
The gold standard for diagnosis is a stereotactic biopsy.[1]
Corticosteroids can lead to marked shrinkage or even the disappearance of such tumors. D/d- sarcoidosis or multiple sclerosis. Histopathological diagnosis is hindered by corticosteroid therapy (CST).[1] Cell arrest and apoptosis from corticosteroid use can cause “ghost” or “vanishing” tumors in up to 50% of cases.[1] Responsiveness to corticosteroids can also be observed in:
- Neurosarcoidosis
- Multiple sclerosis
- Glioblastoma, and
- Vasculitis.[1]
There is an odds ratio of 3.3 for inconclusive biopsy after CST and therefore CST should not be administered before surgery.[1] Non-diagnostic yields following CST range from 33- 57%.[1] Steroids should therefore be withheld prior to diagnostic biopsy owing to their lymphocytolytic effect.[1] Furthermore, most patients however quickly relapse.[1] Close clinical follow-up and serial imaging with the provision of urgent biopsy during re-growth is justified after CST. [1]
High-dose Methotrexate (HD-MTX), a folate antagonist, is the backbone of multimodal chemotherapy.[1] High-dose chemotherapy with autologous stem cell transplantation has a pivotal role in newly diagnosed patients with PCNSL.[1][7]Owing to limited CNS penetration, high doses (≥3.5 g/m) are given.[1] The concomitant use of leucovorin to safeguard bone marrow and systemic organ damage is advised.[7] HD-MTX and Rituximab, a monoclonal antibody against the CD20 antigen, should be part of any induction treatment.[1][7] Intrathecal administration of Rituximab through the ommaya reservoir is recommended among patients with PCNSL with concurrent leptomeningeal involvement.[1]
Due to its high sensitivity to radiation, patients with newly diagnosed central nervous system lymphomas have traditionally received treatment with whole-brain radiotherapy (WBRT). While the initial responses were high, early relapse, as well as radiation-associated neurotoxicity, was common among survivors.[3] Moreover, the median overall survival was 12 to 18 months when WBRT was the sole treatment.[5] With the introduction of high-dose methotrexate (HD-MTX) chemotherapy regimens in the 1970s, improved survival occurred among individuals with CNS lymphoma. When used as a sole treatment, the median overall survival with high-dose methotrexate was 25 to 55 months.
Currently, the mainstay of treatment for individuals with CNS lymphoma is induction chemotherapy that aims for a complete radiographic response (CR), followed by consolidative therapy. The goal of consolidative therapy is to eradicate residual disease and improve overall survival.[3] Induction chemotherapy usually involves some combination of high-dose methotrexate (HD-MTX) with other chemotherapy agents, including temozolomide, cytarabine, etoposide, vincristine, carmustine, ifosfamide, thiotepa, cyclophosphamide. Options for consolidation include high-dose radiation (45 Gy), low-dose radiation (23.4 Gy), and dose-intensive chemotherapy with agents such as carmustine, thiotepa, cyclophosphamide, busulfan, cytarabine, and etoposide. While studies with reduced-dose WBRT as consolidative therapy have shown good progression-free and overall survival rates, a longer follow-up period is required to determine the long-term neuropsychological effects of the radiation. Importantly, in studies that omitted WBRT from consolidative therapy, no survival compromise was shown, and patients had a better neurological outcome.[3][5]
In one study, cytarabine was added to HD-MTX for induction, followed by WBRT for consolidation, with the overall survival at 3 years 46% compared to 32% with HD-MTX monotherapy. In another, thiotepa and rituximab were added to HD-MTX during induction followed by WBRT for consolidation and overall survival was 69% vs. 42% with HD-MTX alone. When using HD-MTX along with procarbazine, vincristine, and rituximab for induction followed by cytarabine and reduced-dose consolidative radiotherapy, the overall survival rate at 3 years was 87% in patients who achieved a complete response (CR) to induction. Also, the median overall survival was not reached at 6 years.[3] The CALGB 50202 trial investigated the use of HD-MTX with oral temozolomide and rituximab (MT-R) for induction, followed by etoposide and cytarabine (EA) for consolidation. 66% of patients achieved a complete response to induction therapy (CR), and the progression-free survival rate was 0.57 at two years.[5] Similarly, the median overall survival was not reached at the 5-year follow-up.
Myeloablative therapy and autologous stem cell transplant are also options for consolidation. In one study, HD-MTX along with cytarabine, thiotepa, and rituximab for induction was followed by myeloablative therapy with high-dose carmustine and thiotepa with autologous stem cell transplant. The overall survival rate at 2 years was 87%. In a similar study, after an HD-MTX-based induction regimen, conditioning therapy was achieved with thiotepa, busulfan, and cyclophosphamide followed by a stem cell transplant. The overall survival rate at 2 years was 81%, and the progression-free survival rate was 79%. At 10 years, the overall survival rate in a similar study was 35%, suggesting that this might be an effective consolidative option, especially for younger patients.[3] The CALGB 51101 was initiated based on the successes of the CALGB 50202 trial and is currently investigating consolidative therapy with etoposide and cytarabine versus myeloablative therapy with carmustine and thiotepa followed by stem cell transplant following, high-dose methotrexate, temozolomide and rituximab (MT-R) based induction.
Unfortunately, more than half of patients with CNS lymphoma experience a relapse, with an average survival of 2 months at that time.[3][13] It is not uncommon for relapses to happen up to 10 years after initial treatment, even though most are seen within 5 years. While the optimal salvage regimen remains investigational, additional HD-MTX, if previously sensitive, along with other central nervous system penetrants such as thiotepa, cytarabine, cytarabine liposome injection, etoposide, and ifosfamide have shown promising results.[5] Additionally, myeloablative therapy followed by stem cell transplant is a good option in younger patients. WBRT if not previously used, remains an effective salvage option with a median overall survival rate of 11 to 19 months.[3] Several new therapeutic agents such as lenalidomide, ibrutinib, buparlisib, nivolumab, pemetrexed, pomalidomide, temsirolimus, and pembrolizumab are currently being investigated.
For the treatment of intraocular lymphoma, one study recommended an HD-MTX and rituximab-based induction, followed by consolidation with cytarabine and etoposide. Intraocular lymphoma also responds well to binocular external beam radiation. Intravitreal rituximab and MTX remains effective option in unilateral disease. But, systemic chemotherapy is the recommendation if the disease is suspected elsewhere within the neuraxis.
Prophylaxis with HD-MTX can be considered for those patients with high-risk systemic non-Hodgkin’s lymphoma to prevent CNS dissemination. High-risk features in these patients include a high international prognostic index score and the presence of extranodal disease, particularly in the testes.[5]
The outcome of the remission can be categorized into the:
- CR-complete response
- Cru-unconfirmed complete response
- PR-partial response
- SD-stable disease, and
- PD-progressive disease.[1]
Current recommendations for the Remission phase of management of PCNSL include:
- If the patient is physiologically fit for intensive therapy-four cycles of MATRix (HD-MTX, cytarabine, thiotepa, rituximab) are recommended.
- If unfit for HD-MTX, options include:
- Regimen incorporating rituximab and an oral alkylating agent such as R-MP [rituximab, MTX, procarbazine]
- Oral chemotherapy (temozolomide)
- Whole-brain radiotherapy, and
- Corticosteroids (dexamethasone).
- Intrathecal chemotherapy is recommended for leptomeningeal disease unfit for systemic therapy.[1]
Consolidation therapy is ideally commenced within 6–8 weeks of the first day of the final induction cycle.[1] Thiotepa/ carmustine with autologous stem cell transplant (ASCT) has a 3- to 5-year overall survival (OS) rate of 70–81%.[1] PRECIS trial and IELSG32 trial have shown differences in progression-free survival (PFS) or OS between WBRT and HDT-ASCT.[1] The reduced risk of neurotoxicity is likely to favor HDT-ASCT in consolidation therapy.[1] High-dose thiotepa-based chemotherapy with ASCT as first-line consolidation should be considered for all eligible patients.[1] WBRT consolidation for consolidation is only advisable for:
- Patients ineligible for HDT-ASCT, and
- Patients with residual disease after thiotepa-based ASCT.[1]
Consolidation phase chemotherapy plus WBRT had no improvement in OS compared with chemotherapy alone during the treatment in the consolidation phase.[7] RT is not required after complete remission following HD-MTX–based primary chemotherapy.[1] Response assessment with contrast-enhanced MRI should be performed 1–2 months after completion of consolidation therapy.[1]
Recommendations for follow-up of the patients:
- At the completion of the therapy then every 3 months for the initial two years.
- Every 6 monthly for the next 3-5 years, and
- Then annually for a total of 10 years. [7]
Mandatory assessments during each follow-up should encompass:
- Thorough medical history and clinical examination
- Cognitive evaluation
- Contrast CT/ MRI brain, and
- Ophthalmologic examination and lumbar puncture if indicated.[1]
Relapsed PCNSL should undergo complete re-staging. Re-staging is not necessary for lesions refractory to first-line therapy.[1]
In systemic non-Hodgkin’s lymphoma, combination chemotherapy is substantially more effective than single-agent therapy.
Differential Diagnosis
The differential diagnosis of CNS lymphoma includes:
- High-grade gliomas
- Metastatic lesions
- Demyelinating diseases
- Granulomatous lesions, and
- Cerebral toxoplasmosis in immunocompromised patients, especially those with AIDs.[1][11]
Toxicity and Adverse Effect Management
Up to 5% of patients develop nephropathy related to high-dose methotrexate use. Adequate hydration, urinary alkalinization, avoidance of penicillin, and other drugs that interact with methotrexate are ways to mitigate this toxicity. Recommendations are to observe a two-day gap between iodinated contrast use for imaging and high-dose methotrexate administration. Leucovorin rescue with escalated dosing strategies, as well as the use of the enzyme carboxypeptidase G2 to facilitate methotrexate clearance via the kidneys, are also effective options.[5]
Leuco-encephalopathy following WBRT is due to the involvement of the neural progenitor cells and is observed in up to 24% of cases and can present as subcortical dementia, gait ataxia, and incontinence.[12]
Complications of systemic non-Hodgkin's lymphoma includes:
- Infectious-due to immunosuppressive agents
- Toxic-metabolic- encephalopathy
- Cerebrovascular-strokes
- Paraneoplastic- motor neuronopathy and necrotizing myelopathy, and
- Treatment-related complications-radiation myelopathy, or cognitive impairment.
Staging
Approximately 4% to 12% of patients originally thought to have primary CNS lymphoma are found to have systemic disease. MRI spine, CT chest, abdomen, and pelvis, bone marrow biopsy, PET imaging, and testis ultrasound in select cases, can be done in the staging process.[3][5] PET imaging can be more sensitive for detecting systemic disease than conventional CT chest, abdomen, and pelvis, and bone marrow biopsy can detect disease not evident in other imaging modalities in the staging process.[3] Baseline levels of LDH, HIV, as well as hepatitis B, and C serology, are often done. Also, since 15 to 25% of patients with CNS lymphoma also have intraocular involvement, an ophthalmologic slit-lamp examination is standard. MRI brain with contrast along with CSF evaluation should also be obtained in patients diagnosed with intraocular lymphoma since 80% of these patients ultimately develop lymphoma in other areas of their central nervous system.[5] Optical coherence tomography and fluorescence angiography can also be options in the diagnosis and staging of intraocular lymphoma.
Prognosis
The 5-year survival rate is almost 40% in recent studies.[7]The 5- and 10-year survival rate for PCNSL is reportedly 30% and 20%.[7]
Two scoring systems have prognostic implications:
- International Extra-nodal Lymphoma Study Group (IELSG)- observed Eastern Cooperative Oncology Group performance status higher than 1; age more than 60 years; elevated serum LDH; elevated CSF protein; and deep cortical lesions as variables prognosticating poor OS.[7]
- Memorial Sloan Kettering Cancer Center's (MSKCC) prognostic score includes age and Karnofsky performance status (KPS).[13]
- Patients less than 50 years- median survival of around 9 years
- Patients more or equal to 50 years and KPS more than 70- median survival of around 3 years.
- Patients at least 50 years old with a KPS less than 70 median- survival of around 1 year.[7]
The positive response to the initial steroid therapy is a good survival marker (117 months vs. 5.5 months in non-responders).[7][12]This however has a high risk of relapse.
BCL-6 expression confers poor survival outcomes.[7] Patients with lymphopenia also have a poor 5-year survival rate (22.3% versus 58.5%).[7]
WBRT alone has shown a median survival of 12 to 18 months and 5-year survival of 18% to 35%.[12] This however has a high risk of neurotoxicity.[12] Radiation Therapy Oncology Group (RTOG) showed that irradiation alone conferred a median survival of only 12 to 18 months with a 5-year survival of only 3–4%. The RTOG studied pre-radiation CHOP followed by whole-brain radiotherapy showed no survival benefits and had significant chemotherapy-related toxicities. The concurrent use of high-dose Methotrexate increased median survival to about 40 months with a 5-year survival rate increase of almost 22%. Patients are vulnerable to leukoencephalopathy seen in 50%-100% of older patients and up to 30% of patients below 60 years of age. Leuco-encephalopathy due to the involvement of the neural progenitor cells is observed in up to 24% of cases and can present as subcortical dementia, gait ataxia, and incontinence.[12] Leukoencephalopathy leads to periventricular white matter changes, ventricular enlargement, and cortical atrophy. [7] Demyelination and hippocampal neuronal loss has been postulated as the probable mechanism.[7] A single battery of psychometric tests has been advocated for the assessment of the same in all patients.[12] Elimination of cranial radiotherapy, therefore, is the goal.WBRT is now only considered for patients with contraindications to chemotherapy or as a salvage therapy in refractory or relapsed cases.[7]
Primary CNS lymphomas usually show rapid and durable responses to Methotrexate. Intravenous MTX alone has shown a CR of 52% and a median overall survival of 55.4 months.[12] Intra-arterial MTX, the CR proportion was 57.8% with a median overall survival of 3.1 years.[12] Disruption of the blood-brain barrier with intra-arterial mannitol, followed by intra-arterial Methotrexate in conjunction with cyclophosphamide, procarbazine, and dexamethasone imparted a median survival of 40 months. However, 31% of them had relapsed and required RT and a high risk of neurotoxicity. Chemotherapy-only trials showed a median OS of 25 to 50 months.[13] Despite high initial response following HD-MTX-based chemotherapy, more than 50% of patients relapse and are to be managed by:
- HD-MTX re-challenge
- High-dose chemotherapy alongside ASCT, or
- WBRT. [7]
Patients with refractory, relapsed, or progressive PCNSL have a median survival of only 4.5 months.[12][13]
Microscopic tumor seedlings residing behind an intact blood-brain barrier account for poor results seen following chemotherapy regimens such as cyclophosphamide, doxorubicin, vincristine, and prednisone or dexamethasone (CHOP or CHOD).
Complications
Several neuropsychological complications, such as gait impairments, memory loss, as well as incontinence, can arise in patients treated with WBRT. These most commonly occur in individuals older than 60 years.[5] Reports also exist of post-traumatic stress disorders in individuals post-treatment. The risk of developing a second malignancy increases in long-term CNS lymphoma survivors, particularly in the younger age groups. Patients with secondary CNS lymphoma would be at an increased risk for radiation-associated cardiovascular disease if applying radiation to the chest. The use of anthracyclines such as doxorubicin has demonstrated cardiotoxicity, and the use of rituximab is associated with an increased risk of progressive multifocal leukoencephalopathy. In post-transplant CNS lymphoma patients, allograft failure is a risk, particularly if the immunosuppressant gets reduced or halted to reconstitute immune function.
Deterrence and Patient Education
Patients require education regarding their disease process as well as potential pitfalls to avoid while undergoing therapy. Methotrexate, when given in high doses, can precipitate in renal tubules as well as cause direct tubular injury. This risk is increased in volume-depleted states as well as with acidic urine. Adequate hydration is of paramount importance. Also, certain drugs such as NSAIDs, penicillins, probenecid, phenytoin, ciprofloxacin, proton-pump inhibitors, and levetiracetam interfere with methotrexate renal clearance, and so should be avoided when possible. Ideally, WBRT should be avoided and if required, low-dose therapy is recommended.
Enhancing Healthcare Team Outcomes
Prompt diagnosis and treatment are pivotal. [7]The interdisciplinary effort of neurology, radiology, neurosurgery, neuropathology, and medical oncology is of paramount importance.[7] The interdisciplinary effort of neurology, radiology, neurosurgery, neuropathology, and medical oncology is of paramount importance.[7] All cases should be discussed at a lymphoma MDT.[1] Definitive treatment should ideally be initiated within 14 days of diagnosis at an established center.[1]
Successful treatment of central nervous system lymphomas requires a collaborative and interprofessional team approach to enhance the patient’s quality of life before, during, and after treatment; this often includes a team of experts in various subspecialties such as:
- Medical oncologists
- Neurologists
- Neuroradiologists
- Pain management specialists
- Physical and occupational therapists
- Psychiatrists
- Neurosurgeons
- Neuropathologists
- Radiation oncologists
- Ophthalmologist
- Social workers
- Spiritual care leaders
- Oncology nurses, and
- Palliative care specialists.
In addition to those listed above, nursing and pharmacy will play crucial roles in managing CNS lymphoma. Nursing will be administering the chemotherapy, which should have input from an oncology specialist pharmacist, who will verify agent selections and verify all dosing while checking against drug-drug interactions, many of which can later therapeutic results, as has been discussed. The nurse should be alert to adverse reactions, as well as noting therapeutic efficacy, and reporting any concerns to the ordering physician. In this way, all these various disciplines can contribute to the collaborative interprofessional team approach to disease management to optimize outcomes. [Level V]
CNS lymphoma has a guarded prognosis; every treatment has significant side effects which add to the morbidity. Even in patients who do respond to treatment, relapse is common. Because of the grim prognosis, a palliative team should have involvement early in the care of these patients. Comfort care and quality of life should not be sacrificed with exhaustive tests and procedures that do not change the prognosis.