Introduction
Skeletal muscle is found throughout the body and functions to contract in response to a stimulus. Skeletal muscle serves many purposes, including producing movement, sustaining body posture and position, maintaining body temperature, storing nutrients, and stabilizing joints. In contrast to smooth and cardiac muscle contraction, most skeletal muscle contraction is under voluntary control, receiving neural inputs allowing conscious control of muscles. Skeletal muscle comprises approximately 40% of the human body weight and contains 50 to 75% of all body proteins.[1]
Issues of Concern
Skeletal muscle disorders typically manifest as muscle weakness. Multiple types of skeletal muscle disorders have been studied, including muscular dystrophies, congenital myopathies, inflammatory disorders, and diseases affecting the neuromuscular junction.
Cellular Level
Skeletal muscle is a highly organized tissue composed of bundles of muscle fibers called myofibers which contain several myofibrils. Each myofiber represents a muscle cell with its basic cellular unit, the sarcomere. Bundles of myofibers form fascicles, and bundles of fascicles form muscle tissue.
Skeletal muscle fibers are striated, multinucleated cells ranging from 10 to 100 micrometers in diameter and many centimeters long. The nuclei are located in the cell's periphery, adjacent to the sarcolemma. The sarcolemma is a tubular sheath that encases and defines each muscle fiber, forming a barrier between extracellular and intracellular compartments. The sarcolemma is comprised of a plasma membrane and a polysaccharide coating that fuses with tendon fibers. Invaginations within the sarcolemma are termed transverse tubules (T tubules), which function as a major location for ion exchange.
Grossly, skeletal muscle fibers are made up of endomysium, perimysium, and epimysium, covering the sarcolemma; each muscle fiber is a layer of connective tissue called the endomysium. Capillaries and nerve tissue are present within the endomysium to supply the individual muscle fibers. Multiple muscle fibers join to form fascicles encased by another connective tissue covering known as the perimysium. The perimysium may surround anywhere from 10 to 100 fascicles. Muscle fascicles are further grouped to form a muscle encased by a fibrous tissue envelope called the epimysium.
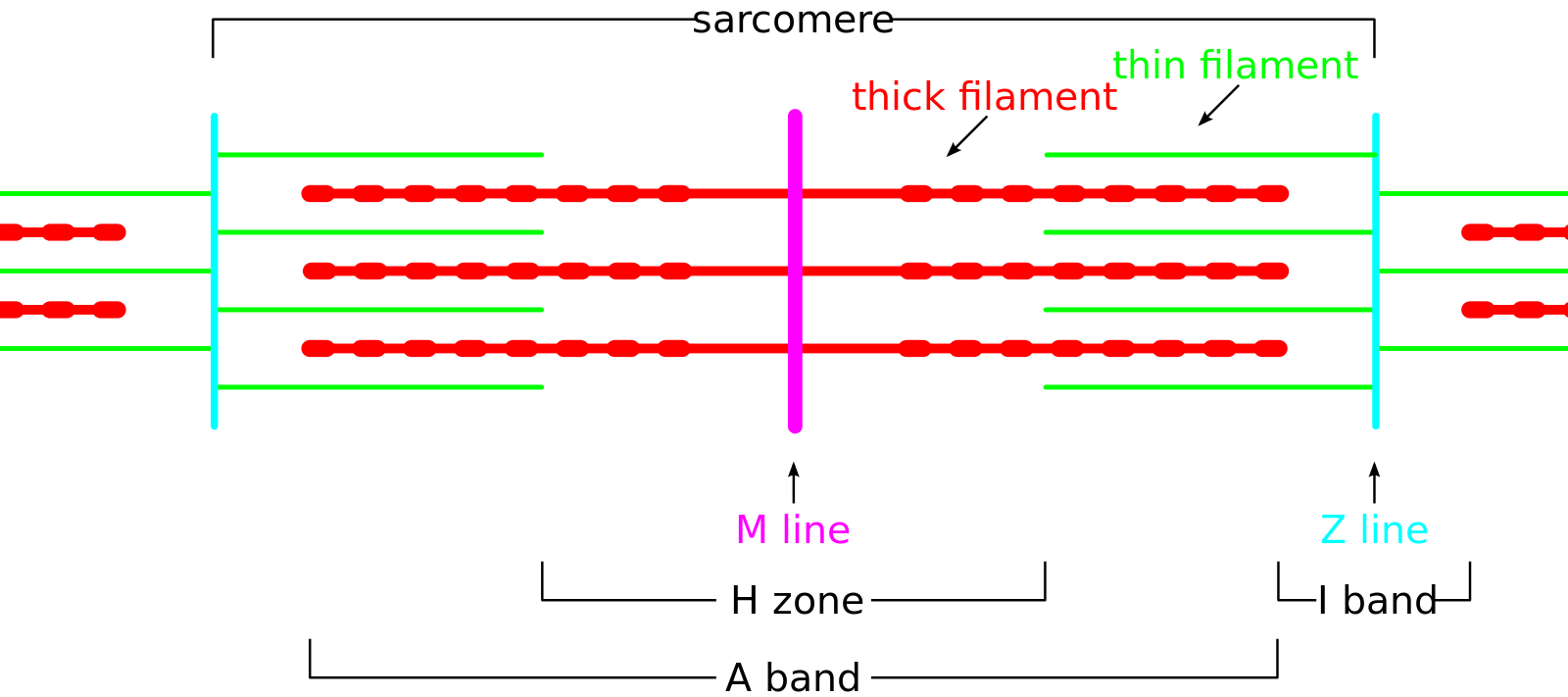
Each muscle fiber is composed of several hundred to several thousand myofibrils. Myofibrils are composed of actin (thin filaments), myosin (thick filaments), and support proteins. The arrangement of actin and myosin gives skeletal muscle its microscopic striated appearance and creates functional units called sarcomeres. When viewed under electron microscopy, sarcomeres are arranged longitudinally and include the M line, Z disk, H band, A band, and I band.
The Z line, or Z disk, is the terminal boundary of the sarcomere, where alpha-actinin acts as an anchor for the actin filaments. The M line is the central-most line of the sarcomere, where myosin filaments are anchored together through binding sites within the myosin filament. The H band contains the M line and is the central region of the sarcomere that contains only myosin filaments. The A band is a larger portion of the sarcomere that contains the entirety of the myosin fibers and includes regions of actin and myosin overlap. The I band covers the terminal regions of two adjacent sarcomeres and contains only actin filaments. The H and I bands shorten with muscle contraction, while the A band remains a constant length.[2]
Actin filaments are double-helical structures, known as filamentous-actin (F-actin), composed of monomeric G-actin units. G-actin exhibits polarity and creates a positive and negative end within the sarcomere, with the positive end situated toward the terminal end of the sarcomere. Tropomyosin is a helical protein that runs along the actin double helix within its groove.[3] Troponin binds tropomyosin by a troponin complex at every seven actin monomer and is composed of troponin-C (Tn-C), troponin-I (Tn-I), and troponin-T (Tn-T). Tn-T binds tropomyosin, Tn-I inhibits actin and myosin binding, and Tn-C binds calcium.[4]
Myosin proteins are composed of two regions: light meromyosin and heavy meromyosin. Light meromyosin binds other light meromyosin regions to anchor myosin at the M line. Heavy meromyosin is further subdivided into two regions; the S-1 portion, or the myosin head, binds actin and contains an ATPase portion, while the S-2 portion is the location of the power stroke.[5]
Support proteins within the sarcomere include titin, desmin, myomesin, C protein, nebulin, and plectin. Plectin tethers the Z disks of adjacent myofibrils to each other. Desmin helps maintain myofibril alignment, connects to the cytoskeleton and other structural elements within the cell, and distributes contractile force. Myomesin and C protein are myosin-binding proteins that function to tether and stabilize myosin at the M line. Titin is found at the Z disk and anchors myosin longitudinally within the sarcomere.
Muscle fiber types can be broken down into three groups. Type I fibers, or slow oxidative fibers, are slow-twitching fibers. They are the smallest fiber type and have a low glycogen content. Type I fibers have a low rate of fatigue, slow contractile speed, and low myosin ATPase activity, making them best suited for endurance types of contraction, such as maintaining posture and marathon running.
Type IIa fibers, or fast oxidative fibers, are fast twitching fibers with a high myosin ATPase activity and an intermediate rate of fatigue. They are best suited for medium-duration and moderate-movement actions like walking and biking.
Type I and IIa fibers are called red fibers, meaning they have a high myoglobin content. They also obtain ATP primarily from oxidative phosphorylation and are composed of many mitochondria and capillaries.
Type IIb fibers, or fast glycolytic fibers, are fast-twitching fibers. They are the largest fibers in diameter due to their high density of actin and myosin proteins. Type IIb fibers contain few mitochondria and are termed white fibers due to their low myoglobin content. These fibers obtain ATP primarily from anaerobic glycolysis, have a high myosin ATPase activity, and have a fast rate of fatigue. They are best suited for short-duration, intense movements such as sprinting and weight-lifting.
Development
The vast majority of muscles are derived from the mesoderm, with skeletal muscles, specifically, derived from the paraxial mesoderm.
Mesodermal cells form myogenic cells, which undergo mitosis to form postmitotic myoblasts. These myoblasts synthesize actin and myosin and fuse to form multinucleated myotubes. Myotubes synthesize actin, myosin, troponin, tropomyosin, and other muscle proteins. These proteins all combine to form myofibrils, the muscle fibers.
The paraxial mesoderm, which eventually forms the skeletal muscles, first divides into segments, or somitomeres, in a craniocaudal fashion. Seven somitomeres form the head and neck muscles and contribute to forming the pharyngeal arches. The remaining somitomeres form 35 pairs of somites for the trunk region. These somitomeres then undergo epithelialization to create groups of epithelial cells.
The mesoderm originates in the somite, with the ventral region of each somite forming the sclerotome or bone-forming cells. The lateral somatic region separates somite clusters from the parietal mesoderm into the primaxial and abaxial domains. The primaxial domain consists of somites around the neural tube; it receives signals for differentiation from the notochord and neural tube and forms the shoulder, back, and intercostal muscles. The abaxial domain receives signals for differentiation from the lateral plate mesoderm and forms the infrahyoid, abdominal wall, and limb muscles.[6]
Organ Systems Involved
Skeletal muscle is found throughout the body, attached to bones via tendons. Skeletal muscle is also present in the tongue, diaphragm, eye socket, and upper esophagus.
Function
The main functions of skeletal muscle are to contract to produce movement, sustain body posture and position, maintain body temperature, store nutrients, and stabilize joints.
From a mechanical standpoint, the primary function of skeletal muscle is to convert chemical energy into mechanical energy, thus generating force and power. From a metabolic point of view, skeletal muscle contributes to basal energy metabolism, serving as a storage site for essential substrates such as carbohydrates and amino acids.
Skeletal muscle also functions to produce body heat. This heat produced is a by-product of muscular activity and is mainly wasted. As a homeostatic response to extreme cold, muscles are signaled to trigger contractions of shivering to generate heat.[7]
Mechanism
Action potentials initiate muscle contraction through excitation-contraction coupling. Excitation-contraction coupling is the mechanism by which neural action potentials convert to cross-bridge cycling, ie, contraction. Action potentials of the motor neuron cause the release of ACh from the neuron terminus at the NMJ or motor endplate. The ACh causes depolarization at the NMJ and transmits the action potential to the muscle fiber. In this process, action potentials travel along the cell membrane and into the T tubules to carry the signal to the interior of the muscle fiber. Depolarization causes a conformational modification in the dihydropyridine receptors of the T tubules, with or without calcium (Ca) influx. This conformational change opens ryanodine receptors on the terminal cisternae of the sarcoplasmic reticulum to release Ca from its storage site in the sarcoplasmic reticulum into the intracellular fluid (ICF), increasing the concentration of ICF Ca by a factor of 10.[8]
The increased ICF concentration of Ca causes a conformational change of the troponin complex by binding troponin C on the actin filament. Each troponin C can bind a maximum of four Ca ions, and binding is cooperative (similar to hemoglobin binding of oxygen). This allows a small change in [Ca] to saturate the troponin C binding sites. The conformational change of troponin C uncovers the myosin-binding sites on actin by pulling tropomyosin out of the way, which begins the cross-bridge cycling that causes skeletal muscle contraction. After excitation and subsequent depolarization of the T tubules ceases, Ca is released from troponin C and sequestered by the sarcoplasmic reticulum in the terminal cisternae through a Ca-ATPase in the membrane of the sarcoplasmic reticulum known as SERCA. Sequestration of Ca allows tropomyosin to cover the myosin-binding sites on actin, causing muscle relaxation.[8]
Cross-bridge cycling is the mechanism by which skeletal muscle contracts. At the beginning of this cycle, myosin is bound tightly to actin in a step termed rigor. In the absence of basic physiologic energy, such as in death, this is a semi-permanent state called rigor mortis. In living tissue, this is a transient state, as ATP binding by the myosin head causes a conformational change of the myosin head that causes the release of the actin-myosin cross-link. After ATP is bound and the cross-link is released, ATP is hydrolyzed to adenosine diphosphate (ADP) and inorganic phosphate, “re-cocking” and moving the myosin head toward the positive end of actin (closer to the ends of the sarcomere).[9]
As long as there is adequate Ca to maintain an uncovered actin-binding site, the myosin head will form a cross-bridge with actin. The release of ADP and inorganic phosphate causes the power stroke where the myosin head moves toward the negative end of actin (toward the center of the sarcomere), displacing the actin filament and shortening the sarcomere. To complete the cycle, ADP is released, and the sarcomere returns to a state of rigor. This cycle repeats as long as Ca is bound to troponin C.[9]
The force of contraction is a summation of the number of motor units recruited and the frequency of action potentials that reach those motor units. The motor unit is defined as a single motor neuron and all the muscle fibers it innervates, with multiple motor units stimulating a single muscle. The number of muscle fibers comprising a motor unit depends on the function of the muscle. Muscles that require fine motor control will involve fewer fibers, whereas larger muscle groups will involve significantly more muscle fibers. Muscles that function over a prolonged period, such as lower back muscles, will asynchronously recruit fibers so that fatigue of individual fibers is spread out over time and space.
Tension on a whole muscle scale results from the additive tension of individual muscle fibers. Recruitment of muscle fibers occurs when an action potential causes the release of a fixed amount of Ca from the sarcoplasmic reticulum, which produces a single muscle twitch followed by sequestration for calcium. Continual stimulation of the muscle by further action potentials causes increased release of calcium and summation of twitches, which allows the muscle to continue contracting. Maximal contraction occurs in a process called tetany when all Ca-binding sites are in use and all myosin-binding sites remain uncovered.[10]
Cleavage of ATP to ADP and inorganic phosphate provides the energy needed for the power stroke mechanism by which skeletal muscle contracts and the reuptake of Ca by the terminal cisternae. ATP stores within the muscle rapidly deplete, so ATP must be regenerated. The first mechanism for ATP generation within the muscle is transferring a phosphate group from creatine phosphate to ADP. Upon the depletion of creatine phosphate stores, ATP production is via the citric acid cycle and the electron transport chain. Oxygen becomes the limiting factor in actively contracting muscle and glycolysis alone, and the subsequent conversion of pyruvate to lactate later becomes the primary source of ATP generation for muscles.[11]
Neuromuscular spindles are present within skeletal muscles and function in proprioception. Intrafusal fibers, or neuromuscular spindles, are interspersed among extrafusal fibers. Intrafusal fibers are composed of two separate cells: the nuclear bag fiber and the nuclear chain fiber. The entire unit functions partly as a muscle, as the fibers can contract, and partly as a sensory receptor for length, tension, and rate of contraction. Primary afferent nerve fibers have annulospiral endings around both the bag and chain fibers and sense muscle length and rate of contraction. Secondary afferent fibers have flower spray endings, mostly on nuclear chain fibers, and function to sense muscle length.[12]
Pathophysiology
Diseases Affecting the Neuromuscular Junction
- Myasthenia Gravis is an autoimmune disorder that results from antibodies to the ACh receptors of the neuromuscular junction. These antibodies prevent ACh from binding and decrease the depolarization transmitted to the muscle cell. As repetitive use consumes ACh stores, lower concentrations of ACh released into the NMJ cannot saturate the binding sites and produce an action potential within the muscle. This presents as a variable weakness in the patient that is worse with use and better with rest. Often, the extraocular muscles are the first muscles affected in the course of the disease. Edrophonium, a short-acting acetylcholinesterase inhibitor, can be used to diagnose myasthenia gravis. When administered, edrophonium prolongs the action of ACh at the NMJ and prevents muscle fatigue for a short time.[13]
- Lambert-Eaton Myasthenic Syndrome (LEMS) is a disorder of the NMJ which may present as a paraneoplastic phenomenon, with more than half of the cases associated with small cell lung cancer (SCLC). The primary clinical manifestation is muscle weakness. The pathology is due to the generation of antibodies against voltage-gated Ca channels on presynaptic nerve terminals, leading to a decrease in the neurotransmitter ACh. In addition to muscle weakness, those with LEMS may present with oculobulbar weakness, dysphagia and dysarthria, and autonomic dysfunction. Respiratory failure may infrequently occur in the late stages of LEMS.[14] Postexercise or post-activation facilitation is a phenomenon seen in LEMS associated with an improvement in muscle weakness following exertion. Repetitive muscle contractions cause an increased influx of Ca in the presynaptic membrane, facilitating the release of ACh by binding with multiple vesicles. This effect is temporary, as the mitochondria eventually clear the excess calcium.
- Toxins may also affect excitation-contraction coupling at the NMJ. Botulinum toxin, produced by C. botulinum, inhibits ACh release from the presynaptic neuron at the NMJ, preventing skeletal muscle excitation and causing flaccid paralysis. Tetanospasmin, a neurotoxin released by C. tetani, prevents relaxation through blockage of inhibitory neurotransmitter release by interneurons that synapse at the NMJ, which leads to spastic paralysis.[15]
Muscular Dystrophies
Muscular dystrophies include more than 30 inherited conditions that cause permanent muscle weakness. The two most common and well-known forms of muscular dystrophy include Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD).
- Duchenne Muscular Dystrophy (DMD) is an X-linked inherited neuromuscular disorder caused by mutations in the dystrophin gene. This disease is characterized by progressive muscle wasting and weakness due to the absence of dystrophin protein that causes skeletal and cardiac muscle degeneration. DMD affects approximately 1 in 3,600 male births worldwide, with clinical signs absent at birth. The average age of diagnosis, and first appearance of symptoms, is around the age of 4. DMD is a fast-progressing disease, with patients relying on a wheelchair and developing severe cardiomyopathy at approximately ten years of age. Death typically occurs due to cardiac and respiratory complications.[16]
- Becker Muscular Dystrophy (BMD) is described as a milder form of DMD, with an incidence of 1 in 18,518 male births. BMD typically presents later than DMD, between the ages of 5 and 15, with a slower clinical progression. Typically, those affected by BMD show an onset of symptoms after age 30 and may remain ambulatory into their 60s. Despite milder skeletal muscle involvement, heart failure from dilated cardiomyopathy is a common cause of morbidity and the most common cause of death in BMD patients.[17]
Idiopathic Inflammatory Myopathies (Myositis)
- Dermatomyositis (DM) presents with symmetric proximal muscle weakness developing over weeks to months in conjunction with erythematous changes, which may precede or follow the myopathy. Erythematous changes include a heliotropic rash, eyelid edema, Gottron papules at extensor surfaces, and subcutaneous calcifications. Myalgia is not typically seen, but patients may develop dysphagia, dysarthria, and interstitial lung disease. DM shows an increased prevalence in females and older patients.[18]
- Polymyositis (PM) presents with subacute onset of proximal muscular weakness, most pronounced in the pelvic girdle and shoulders, and a marked elevation of creatine kinase (CK). The neck flexors are also commonly affected, and in some cases, the neck extensors. A biopsy showing cellular infiltrates composed of macrophages and cytotoxic CD8+ T cells distinguish PM from DM.[19]
- Necrotizing Myopathy (NM) is clinically indistinguishable from PM, with progressive, symmetrical weakness of the proximal muscles of the arms and legs. Myalgias occur in up to 80% of patients, and dysphagia and dysarthria may develop in severe cases. NM shows a prevalence for males, with a mean age of onset in the 5th decade. A biopsy is required to diagnose NM, showing scattered necrotic muscle fibers with sparse inflammatory cells surrounding the necrosis, predominant macrophages, and a few lymphocytes (CD4+ and CD8+ T cells).[20]
- Inclusion Body Myositis (IBM) is the most frequently acquired myopathy after age 50. IBM is a slow-developing disease, which typically affects flexion of the hand and fingers and knee extension. Dysphagia is typical, and at least minor swallowing difficulties are observed in up to 80% of affected individuals. Dysphagia may precede weakness in the arms and legs. Eventually, patients typically become wheelchair-dependent, but life expectancy is normal.[21]
Rhabdomyolysis is a direct injury to skeletal muscle architecture, leading to the release of intracellular contents, such as electrolytes and myoglobin, into the extracellular space. The insults leading to skeletal muscle injury are numerous and beyond the scope of this article, but some examples include overuse (eg, marathon running), compartment syndrome, and using prescription, over-the-counter, and illicit drugs. The cellular insult directly results from releasing large amounts of ionized calcium from the terminal cisternae, which activates degradation processes. The downstream effects of rhabdomyolysis can be systemic and life-threatening, with damage to the skeletal muscle leading to the release of breakdown products that impede kidney function and, in severe cases, lead to acute renal failure requiring dialysis.[22]
Atrophy of skeletal muscle may result from many things, including disuse, denervation, systemic illness, chronic glucocorticoid use, and malnourishment. While the pathways may differ, in all these cases, atrophy is caused by increased proteolysis mediated by the ubiquitin system and decreased protein synthesis, which reduces muscle mass by reducing the diameter of individual muscle fibers.[23]
Clinical Significance
Skeletal muscles allow humans to move and perform daily activities and play an essential role in respiratory mechanics and maintaining posture and balance. Various medical conditions can occur as a result of skeletal muscle dysfunction. These diseases include myopathies, dysphagia, ataxia, weakness, tremors, tendon ruptures, and more. Clinically, patients may present with everything from muscle cramps to genetic abnormalities affecting their skeletal muscles.
Appreciation of the pathology behind skeletal muscle diseases has its basis in understanding normal physiology. Of note, it is important to recommend exercise to patients to develop muscle strength. Building and maintaining muscle strength is essential for bone health, balance, flexibility, posture, and overall health.