Continuing Education Activity
21-hydroxylase deficiency is an autosomal recessive disorder caused by a deficiency in one of the enzymes required for the synthesis of cortisol in the adrenal glands. 21-Hydroxylase deficiency is the most common cause of congenital adrenal hyperplasia but can be a challenge to diagnose and treat. This activity reviews the use of glucocorticoid and mineralocorticoid replacement as the mainstays of treatment and highlights the role of a well-coordinated interprofessional care team in improving outcomes for patients with congenital adrenal hyperplasia.
Objectives:
- Review the pathophysiologic basis of 21-hydroxylase deficiency.
- Describe the expected history and physical findings for a patient with 21-hydroxylase deficiency.
- Outline the recommended screening tests to evaluate for 21-hydroxylase deficiency.
- Summarize the need for an interprofessional team approach to caring for a patient with 21-hydroxylase deficiency.
Introduction
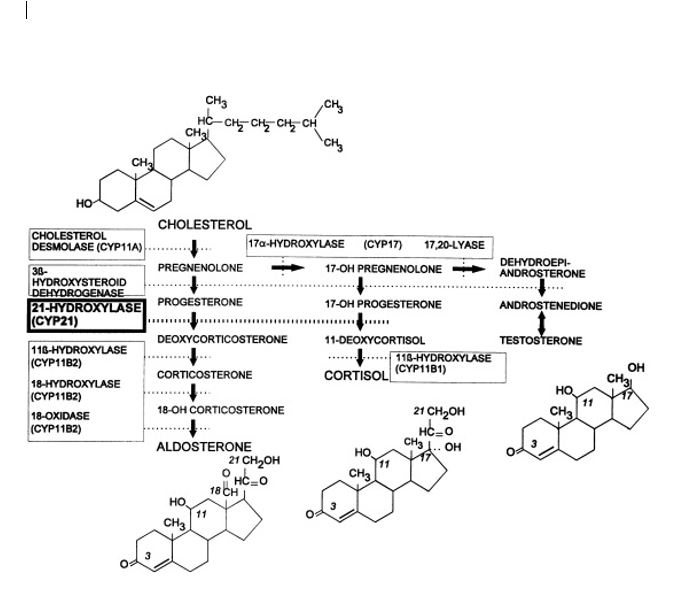
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder, caused by the deficiency of one of the enzymes required for the synthesis of cortisol in the adrenal glands. 21-hydroxylase deficiency is the most common cause of congenital adrenal hyperplasia (more than 90% of the cases). Glucocorticoid and mineralocorticoid replacement are the mainstays of treatment.[1][2][3]
Prenatal diagnosis and treatment of affected females are very important, to minimize genital virilization. Because 21-hydroxylase deficiency is often undiagnosed in affected males until they have severe adrenal insufficiency, all US states and many other countries have instituted newborn screening programs that measure 17-hydroxyprogesterone concentration). Newborn screening can detect almost all infants with classic CAH and some infants with nonclassic CAH. Although false-negative results are uncommon, false-positive results are usually seen in premature infants; therefore, serial measurements of 17-hydroxyprogesterone are advised for premature infants. A positive newborn screening test for CAH must be confirmed by a second plasma sample (17-hydroxyprogesterone), and serum electrolytes should be measured.
This activity highlights the diagnosis and treatment of 21 hydroxylase deficiency.
Etiology
CAH is an autosomal recessive disorder. The activity of 21-hydroxylase is mediated by cytochrome p450c21, found in the endoplasmic reticulum.[4][5][6]
The 21-hydroxylase genes (CYP21) lie within the class III region of the human major histocompatibility complex on chromosome 6. The CYP21 gene structure contains both CYP21 and a pseudogene (CYP21P). CYP21 is the active gene. More than 90% of mutations causing 21-hydroxylase deficiency are recombination between CYP21 and CYP21P.
Multiple gene defects have been found. Several mutations entirely prevent the synthesis of a functional protein; whereas, others are missense mutations that yield enzymes with 1% to 50% of normal activity. Disease severity correlates well with the mutations carried by an affected individual; for example, patients with a salt-wasting disease usually carry mutations on both alleles that destroy enzymatic activity.
Patients are frequently compound heterozygotes for different types of mutations (i.e., one allele is less-severely affected than the other), in which case the severity of disease expression is largely determined by the activity of the less-severely affected of the 2 alleles.
Molecular genetic testing of the CYP21 gene is available and can detect common mutations and deletions in the various forms in up to 95% of affected individuals.
Epidemiology
Congenital adrenal hyperplasia occurs among all races. The worldwide incidence of classical 21-hydroxylase-deficient CAH is 1 in 15,000 to 20,000 births. Approximately 70% of the affected infants have the salt-wasting form, and 30% have the simple virilizing form.[7][8]
Nonclassic congenital adrenal hyperplasia has a prevalence of approximately 1 in 1000 in the general population but occurs most frequently in specific ethnic groups such as Ashkenazi Jews and Hispanics.
Pathophysiology
Steroid 21-hydroxylase (CYP21, P450c21) is a cytochrome e P-450 enzyme located in the endoplasmic reticulum. It hydroxylates 17-hydroxyprogesterone to 11-deoxycortisol, a precursor of cortisol and also hydroxylates progesterone to deoxycorticosterone, a precursor of aldosterone. Both hormones (cortisol and aldosterone) are deficient in the most-severe, ’salt-wasting’ form of the disease.[9]
Due to the loss of this enzyme function, patients with a 21-hydroxylase deficiency cannot synthesize cortisol efficiently, and as a result, the ACTH levels are high, leading to hyperplasia of the adrenal cortex and overproduction of cortisol precursors. Some of these precursors are used for the synthesis of sex steroids, which may cause signs of androgen excess, including ambiguous genitalia in newborn girls and rapid postnatal growth in both sexes. Aldosterone deficiency may lead to salt wasting with consequent failure to thrive, hypovolemia, and shock.
History and Physical
Classification
Classic 21-Hydroxylase Deficiency
Salt-wasting (severe form, with a defect in cortisol and aldosterone biosynthesis)
Approximately 75% of patients with classic 21-hydroxylase deficiency have the salt-wasting type. It is the most severe form of the disease, and it is most often associated with large gene deletions or intron mutations that result in no enzyme activity.
The biochemical and clinical abnormalities of the classic form manifest both prenatally and postnatally. Physicians recognize the condition in infant females in the neonatal period because of ambiguous genitalia compared with that of infant males. Males have normal genitalia and, in the case of the salt-wasting form, they can present with nonspecific symptoms like vomiting, dehydration, and poor feeding at ages 1 to 3 weeks. Hence, the diagnosis in boys can be delayed or missed.
Since aldosterone regulates sodium homeostasis, untreated patients will have excessive renal sodium excretion resulting in hypovolemia and hyperreninemia. These patients cannot excrete potassium efficiently and are prone to hyperkalemia, especially in infancy. In addition, accumulated steroid precursors may directly antagonize the mineralocorticoid receptor and exacerbate mineralocorticoid deficiency, particularly in untreated patients. Progesterone is well known to have anti-mineralocorticoid effects.
Cortisol deficiency contributes to poor cardiac function, poor vascular response to catecholamine, a decreased glomerular filtration rate, and increased secretion of antidiuretic hormone. Cortisol and aldosterone deficiency together cause hyponatremic dehydration and shock in inadequately treated patients.
Since high levels of glucocorticoids are needed for normal development of the adrenal medulla, as well as, for expression of the enzymes required to synthesize catecholamines, patients with salt-wasting type may also have catecholamine deficiency, further increasing the shock.
Females are exposed to high systemic levels of adrenal androgens from week 7 of gestation. Thus, they have ambiguous genitalia at birth: a large clitoris, rugated and potentially fused labia majora, and a common urogenital sinus instead of separate urethra and vagina. The uterus, fallopian tubes, and ovaries are normally formed, but there is no development of Wolffian ducts.
Postnatally, in untreated or inadequately treated patients, long-term exposure to high sex hormones promotes rapid somatic growth and advanced bone age. Linear growth is affected, even with close therapeutic monitoring. Pubic and axillary hair may develop early. Clitoral growth may continue in girls. Young boys may have penile growth despite having small testes. Long-term exposure to androgens may activate the hypothalamic-pituitary-gonadal axis, causing centrally mediated precocious puberty.
Girls may present with oligomenorrhea or amenorrhea in adolescence. As surgical, medical, psychological treatments have improved, more women with 21-hydroxylase deficiency, have completed pregnancies and giving birth.
The prevalence of testicular adrenal rests in boys with classic CAH aged 2 to 18 years varies from 21% to 28%. These so-called testicular adrenal rest tumors are benign, often related to suboptimal therapy, and usually, a decrease in size after optimization of glucocorticoid therapy. Testicular masses in boys with classic CAH are usually bilateral and smaller than 2 cm in diameter and therefore not palpable but detectable by ultrasound.
Simple Virilizing (normal aldosterone biosynthesis)
Approximately 25% of patients with classic 21-hydroxylase deficiency present with simple virilization without salt wasting. The simple virilizing form most commonly results from point mutations that lead to amino acid substitution, causing low but detectable enzyme activity resulting in adequate aldosterone secretion, but decreased levels of cortisol.
Females present at birth with ambiguous genitalia. Without newborn screening, affected boys are diagnosed in childhood when signs of androgen excess develop. Later diagnosis is associated with greater difficulty in achieving hormonal control, and short stature.
Non-classic 21-Hydroxylase Deficiency (mild form)
The nonclassic or late-onset form is more common, occurring in 0.1% to 0.2% in the general white population and 1% to 2% among Ashkenazi Jews. Females with the nonclassic form may be compound heterozygotes with a classic mutation and variant allele or heterozygotes with two variant alleles, allowing 20% to 60% of normal enzymatic activity.
Compound heterozygote females have a less severe phenotype, and clinical presentation varies. Females may present at any age but usually not younger than 6 months. Heterozygote females may have mild biochemical abnormalities but no clinically important endocrine disorder.
Patients with nonclassic form, have normal levels of cortisol and aldosterone at the expense of mild to moderate overproduction of sex hormones precursors. Newborn screening can detect nonclassic cases, but most are missed because of relatively low baseline levels of 17 hydroxyprogesterone.
Hirsutism is the single most common symptom at presentation, followed by oligomenorrhea and acne. Thus, nonclassic 21-hydroxylase deficiency and polycystic ovarian syndrome may present in similar ways.
Evaluation
The screening workup should include:
- Newborn screening programs check for 21-hydroxylase deficiency
- 17-hydroxyprogesterone will be very high (usually greater than 1000 ng/dL) in a patient with the classic form
- Hyperkalemia, hyponatremia, low aldosterone, and high plasma renin activity (PRA), particularly the ratio of PRA to aldosterone, are markers of impaired mineralocorticoid synthesis
- An ACTH stimulation test should be performed to evaluate adrenal function and differentiate among the various potential enzymatic defects. Administration of 0.25 mg of cosyntropin (a synthetic ACTH) provides a pharmacologic stimulus to the adrenal glands, maximizing hormone secretion.
- A full adrenal profile, including measurement of 17-hydroxyprogesterone (17-OHP), cortisol, deoxycorticosterone, 11-deoxycortisol, 17-hydroxypregnenolone, dehydroepiandrosterone (DHEA), and androstenedione, should be obtained immediately before and 60 minutes after cosyntropin administration.
- Nomograms are available for interpreting the results
- In an infant with ambiguous genitalia, do karyotype to establish the chromosomal sex
- The pelvic ultrasound should be done to check for uterus or associated renal anomalies
- A bone age study is helpful in patients with precocious pubic hair
- In patients with signs of acute adrenal failure, CT of the adrenal glands can be done to exclude adrenal hemorrhage
- Urogenitography for defining the anatomy of the internal genitalia
Treatment / Management
Acute Adrenal Crisis
- Is a medical emergency
- Initial management should be fluid resuscitation: Intravenous (IV) bolus of isotonic sodium chloride solution (20 mL/kg). Repeated boluses may be needed.
- Administer dextrose if the patient is hypoglycemic and must be rehydrated with fluid after the bolus dose to prevent hypoglycemia
- Stress doses of hydrocortisone (100 mg/m2 per day) are vital in the management and should be given as early as possible, concomitant with IV fluid treatment.
- Central access and vasopressors, along with higher glucose concentrations, may be required in profoundly ill patients.
- Life-threatening hyperkalemia may require additional therapy with potassium-lowering resin, IV calcium, insulin, and bicarbonate.
Positive Newborn Screen
Newborn screening for CAH is routinely performed in all 50 US states and at least 40 other countries.[4][10][11]
- A positive newborn screening test for CAH must be confirmed by a second plasma sample (17-hydroxyprogesterone), and serum electrolytes should be measured.
- After the confirmatory blood sample is obtained, treatment doses of glucocorticoid and mineralocorticoid should be initiated in all infants in whom CAH is a consideration, to prevent the potentially life-threatening manifestations of an adrenal crisis.
- If the physician chooses not to initiate treatment while awaiting confirmatory steroid hormone measurements, serum electrolytes should be measured daily.
- A pediatric endocrinologist should manage these patients.
Long-Term Management
The goal of therapy is to reduce excessive androgen secretion by replacing the deficient hormones. Proper treatment prevents adrenal crisis and virilization, allowing normal growth and development, normal pubertal development, sexual function, and fertility.
Glucocorticoids
- Cortisol replacement: Oral hydrocortisone, in three divided doses of 10 to 20 mg/m2 per day.
- Patients with classic 21-hydroxylase deficiency require long-term glucocorticoid treatment to inhibit excessive secretion of CRH and ACTH and reduce the abnormally high serum concentrations of adrenal androgens.
- Hydrocortisone is the treatment of choice because of its short half-life and minimal growth-suppressive effect.
- The efficacy of treatment is best assessed by monitoring ACTH, 17-OHP, DHEA, and androstenedione. A target 17-OHP range of 500 to 1000 ng/dL, although still higher than normal, helps to avoid the adverse effects of overtreatment. Children also should have an annual bone age radiograph and careful monitoring of linear growth
- Older children and adolescents, where growth is complete, may be treated with prednisone (5 to 7.5 mg daily in 2 divided doses ) or once-daily dexamethasone (0.25 to 0.5 mg).
Mineralocorticoids
- Infants born with the salt-wasting form of 21OHD require replacement with mineralocorticoids. Fludrocortisone (usually 0.1 to 0.2 mg, but occasionally patients require up to 0.4 mg per day) and sodium chloride (1 to 2 g, each gram of sodium chloride contains 17 mEq of sodium)
- The sodium content of human milk or most infant formulas are about eight mEq/L, and is insufficient to compensate for sodium loses in these infants.
- Plasma Renin activity levels may be used to monitor the effectiveness of mineralocorticoid and sodium replacement. Hypotension, hyperkalemia, and elevated renin levels suggest the need to increase the dose, whereas hypertension, tachycardia, and suppressed PRA production are clinical signs of overtreatment.
- Excessive increases in fludrocortisone dosage also may retard growth.
Surgical Care
Infants with ambiguous genitalia require a surgical evaluation and, if needed, plans for corrective surgery. Risks and benefits of surgery should be fully discussed with parents of affected females.
Significantly virilized females usually undergo surgery before 1 year of age. If there is severe clitoromegaly, the clitoris is reduced, with partial excision of the corporal bodies and preservation of the neurovascular bundle. Vaginoplasty and correction of the urogenital sinus usually are performed at the time of clitoral surgery. Revision in adolescence is often necessary.
Bilateral adrenalectomy for CAH is controversial. May be considered only in select cases that have failed medical therapy, especially in rare cases of adult females with salt-wasting CAH and infertility. The risk for noncompliance must be considered before surgery.
Differential Diagnosis
- CAH due to 11- beta-hydroxylase Deficiency
- CAH due to 3 beta-hydroxysteroid dehydrogenase deficiency
- Five-alpha-reductase deficiency
- Adrenal hypoplasia
- Adrenal insufficiency
- Androgen insensitivity syndrome
- Bilateral adrenal hemorrhage
- Other causes of hyperkalemia/hyponatremia
- Hypertrophic pyloric stenosis
- Obstructive uropathy
- Polycystic ovarian syndrome
- Defects in testosterone synthesis
- Denys-Drash syndrome
- Disorders of gender development
- Familial glucocorticoid deficiency
Prognosis
Children who have CAH often are tall in early childhood, but ultimately are short in adulthood. Recent data suggest that patients born with CAH are about 10 cm shorter than their parentally based targets. Advanced bone age and central precocious puberty due to androgen excess causing early epiphyseal fusion are the primary factors. In addition, the treatment of CAH with glucocorticoids can suppress growth and diminish the final height. Experimental treatment with growth hormone and luteinizing hormone-releasing hormone analog (to hold off puberty) are reported to lead to an average height gain of 7.3 cm.[12][13][14]
The influence of prenatal sex steroid exposure on personality is controversial. More consistent evidence regarding the effects of androgens comes from gendered play activities of young children.
Most children with CAH manifest normal neuropsychological development. Moreover, despite a tendency toward male gender role behavior and homoerotic fantasy, most girls with CAH identify as females and exhibit a heterosexual preference. Both male and female patients are fertile but have reduced fertility rates. This consequence is due to biological, psychological, social, and sexual factors.
Bone density is reported to be normal in most patients. The prevalence of metabolic abnormalities such as obesity, insulin resistance, dyslipidemia, and the polycystic ovarian syndrome has been reported to be high due to the diseases themselves or glucocorticoid treatment
This disease and treatment complications and long-term consequences are challenging for practitioners. Multiple subspecialty professionals should be involved in management. Gene therapy shows the potential for a CAH cure.
Complications
Complications of congenital adrenal hyperplasia are common. If the patient does not get enough glucocorticoids, he or she can develop adrenal insufficiency and further virilization in the virilizing forms. If a patient receives excessive glucocorticoids, he or she can develop growth failure, obesity, striae, hypertension, hyperglycemia, and cataracts.
The complications of excess mineralocorticoid administration include hypertension and hypokalemia.
Aldosterone deficiency may lead to salt wasting with consequent failure to thrive, hypovolemia, and shock.
Deterrence and Patient Education
Patient and family education on proper treatment, especially compliance with medical therapy is essential for patients affected with this disease. Over and under treatment with glucocorticosteroids can have profound effects on the growth and health of these patients. Caregivers and patients themselves when they are older need to be counselled on proper treatment and when to seek medical care.
Pearls and Other Issues
- Both molecular genetic testing of the fetus and prenatal treatment of mothers with dexamethasone are available.
- Dexamethasone must be given before the seventh to eighth week of gestation to suppress the fetal pituitary-adrenal axis before virilization occurs.
- In a patient with ambiguous genitalia, the diagnosis is not difficult; however, in affected males with no symptoms, newborn screening may be lifesaving. Without it, the diagnosis can be missed until the patient is in an acute adrenal crisis.
- The objective of the treatment of CAH is to prevent adrenal crisis and virilization and to achieve normal growth, pubertal development, sexual function, and fertility. Both male and female patients are fertile but have reduced fertility rates. This consequence is due to biological, psychological, social, and sexual factors.
- The prevalence of metabolic abnormalities such as obesity, insulin resistance, dyslipidemia, and the polycystic ovarian syndrome has been reported to be high due to the diseases themselves or glucocorticoid treatment.
Enhancing Healthcare Team Outcomes
Congenital adrenal hyperplasia is a relatively rare autosomal recessive disorder, but the diagnosis carries significant mental and physical morbidity and mortality. The condition is best managed by an interprofessional team that includes a geneticist, endocrinologist, pediatrician, nurse, and a mental health counselor. Prenatal diagnosis and treatment of affected females is very important to minimize genital virilization. Because 21-hydroxylase deficiency is often undiagnosed in affected males until they have severe adrenal insufficiency, all US states and many other countries have instituted newborn screening programs that measure 17-hydroxyprogesterone concentration). Newborn screening can detect almost all infants with classic CAH and some infants with nonclassic CAH.[15]