Continuing Education Activity
Corneal dystrophies are a collection of genetic diseases that affect corneal transparency and refraction due to increased deposition of abnormal material. This activity will review the classification of these diseases, their clinical presentations, diagnostic approaches, and clinical management by an interprofessional team.
Objectives:
- Describe the corneal dystrophies based on the affected anatomical layer.
- Outline various corneal dystrophies based on genetic etiology, history, physical exam findings, and laboratory diagnostics.
- Review the management of corneal dystrophies, from non-invasive treatments to corneal transplantation, as well as the risks and complications of surgical intervention.
- Summarize an interprofessional team-based approach to the management of corneal dystrophies for early diagnosis, proper treatment and improved outcomes.
Introduction
Corneal dystrophy (CD) is most recently defined as a collection of rare hereditary non-inflammatory disorders of abnormal deposition of substances in the cornea.[1] CD was coined in 1890 by Arthur Groenouw and Hugo Biber, and the efforts of Ernst Fuchs, Wilhelm Uhthoff, and Yoshiharu Yoshida solidified the foundation of the understanding of these diseases.[1] As proposed in 2015 by the International Classification of Corneal Dystrophies (IC3D), CD is sub-classified by the anatomic location affected: epithelial/subepithelial, epithelial-stromal, stromal, and endothelial dystrophies.[1] Discoveries and unique case studies continue to broaden our understanding and classification of these diseases; therefore, it is difficult to categorize every single dystrophy solely into these four major labels.
The objective of this article is to present an overview of the evaluation and management for the most prominent and understood variants of CD.[1] Highlights of these dystrophies will be discussed. However, further in-depth discussion on these dystrophies will be in separate StatPearls articles.
Patients with CD can be asymptomatic, but if symptoms occur, they typically experience bilateral visual acuity loss, typically in the form of irregular astigmatism. Depending on the corneal layer affected, patients may also manifest with photophobia, dry eyes, corneal edema, and recurrent corneal erosions, especially with epithelial-based CD, which causes considerable pain. Symptoms can begin at any age, depending on the diagnosis.[2] Treatment can range from conservative measures to corneal transplantation.[1][2]
CD is a significant but rare ocular disease. The genetic component of this disease is important for patients to understand, especially for affected patients involved with family planning. As we begin to understand genetics in greater detail, better evaluation and treatments for CD will come to fruition.
The objective of this article is to present an overview of the general evaluation and management for the most prominent and understood variants of CD.[1] Highlights of these dystrophies will be covered, but the author intends to elaborate on these dystrophies separately in other StatPearls articles. The variants of CD based on their new anatomic classifications in IC3D are[1]:
Epithelial and subepithelial dystrophies
- Epithelial basement membrane corneal dystrophy (EBMCD), also previously known as map-finger-dot dystrophy, Cogan microcystic dystrophy, and anterior basement membrane dystrophy.[1][2]
- Epithelial recurrent erosion dystrophies (EREDs) which includes Franceschetti corneal dystrophy, dystrophia smolandiensis, and dystrophia helsinglandica[1]
- Subepithelial mucinous corneal dystrophy (SMCD)
- Meesmann corneal dystrophy (MECD) also known as juvenile epithelial corneal dystrophy[1][3]
- Lisch epithelial corneal dystrophy (LECD)
- Gelatinous drop-like corneal dystrophy (GDLD)
Epithelial-Stromal Dystrophies (still included under epithelial and subepithelial dystrophies)[1]
- Lattice corneal dystrophy (LCD), with its subtypes: type I (TGFBI mutation) and type II (familial amyloidosis Finnish type),[1] including LCD variants
- Granular corneal dystrophy (GCD), types I and II (Avellino-type)[2]
- Reis-Bückler’s corneal dystrophy (RBCD)
- Thiel-Behnke corneal dystrophy (honeycomb dystrophy)[4] (TBCD)
Stromal dystrophies
- Macular corneal dystrophy (MCD)
- Schnyder corneal dystrophy (SCD)
- Congenital stromal corneal dystrophy (CSCD)
- Fleck corneal dystrophy (FCD)
- Posterior amorphous corneal dystrophy (PACD)
- Pre-Descemet corneal dystrophy (PDCD)
- Central cloudy dystrophy of francois (CCDF)
Endothelial Corneal Dystrophies
- Fuchs endothelial corneal dystrophy (FECD)
- Posterior polymorphous corneal dystrophy (PPCD)
- Congenital hereditary endothelial dystrophy (CHED)
- X-linked endothelial corneal dystrophy (XECD)
Etiology
Clinicians need to distinguish corneal degeneration from corneal dystrophies. Corneal degeneration is a broad term for afflictions that cause alterations to the corneal structure due to a multitude of factors, such as trauma, infection, increasing age, and other environmental factors. Corneal dystrophy is a disorder distinguished from general corneal degeneration by a genetic etiology, either inherited or through de novo mutation.[2] Historically, when an understanding of genetics was limited, CD was differentiated by its bilateral, symmetric, insidious presentation that seemed not to be related to environmental factors.[1][5] Additionally, corneal dystrophies tend to affect vision more than corneal degeneration.
The IC3D categorized CD into four different categories, based on our genetic understanding of each disease variant: category 1 identifies a CD with the mutated gene(s) identified, category 2 identifies the locus of the mutation, category 3 is a CD without a gene or locus identified, and category 4 is reserved for new or suspicious CD.[1][6] Many dystrophies are well understood genetically and are, therefore, in category 1. Lisch epithelial CD and XECD are in category 2.[4] Others, such as ERED and posterior amorphous CD (category 3) and subepithelial mucinous CD (category 4), are not yet well understood genetically.[4]
Many corneal dystrophies are inherited in an autosomal dominant pattern.[1][4][7] There are autosomal recessive CDs as well, and studies suggest that CD can be a result of consanguineous families.[4] Lisch epithelial CD and XECD are X-linked dominant diseases.[1] These X-linked disorders may be associated with other diseases that are concomitantly inherited, such as keratosis pilaris, or congenital, inflammatory papules that result in hair loss.[8][9]
While many genes could be affected and lead to corneal dystrophy, some genes have been confirmed:
Transforming Growth Factor, Beta Induced (TGFBI)
This gene is altered in epithelial-based dystrophies such as type I LCD, GCD types I and II, EBMCD, Reis-Bücklers CD, and Thiel-Behnke CD.[2][4][10][11] Also known as BIGH3 or keratoepithelin, this gene is found on chromosome 5, and the function of the protein it produces is to link to collagens and provide support in the corneal extracellular matrix.[12] Some variants of LCD are autosomal recessive, all presenting with similar symptoms as the other types of LCD, but with different mutations on the keratoepithelin gene.[13][14]
Gelsolin: Type II LCD (Finnish type) is caused by a mutated form of gelsolin. This protein normally functions in removing excess actin filaments from plasma, but that can cause systemic amyloidosis when mutated.[15][16] This leads to amyloid deposition in vascular, skin, and nerve tissue, as well as the cornea.[17]
KRT: Meesmann CD is an autosomal dominant disease caused by a mutation in the Keratin gene, specifically KRT3 and KRT12, which normally produce intermediate filaments inside cells to provide structure to keratocytes.[18] The dysfunction of keratin 3 and 12 weakens epithelial cells to the point of discohesion.[3][4][18][19]
UBIAD1: Schnyder CD is caused by a mutation in the UbiA prenyltransferase domain-containing protein 1 gene, which is involved in vitamin K and coenzyme Q metabolism.[20]
TACSTD-2: An autosomal recessive mutation of tumor-associated calcium signal transducer-2 is found in gelatinous drop-like dystrophy.[2][21]
CHST6: Carbohydrate sulfotransferase 6 is found in MCD and inherited in an autosomal recessive fashion.[22] The gene encodes for a sulfate-transferring protein.[2]
SLC4A11: Found in FECD, late-onset, as well as congenital hereditary endothelial dystrophy, this gene produces a bicarbonate transporter protein.[4][23][24]
ZEB1: Zinc finger E-box-binding homeobox 1, formerly known as TCF8, leads to Posterior Polymorphous CD, as well as FECD, and is responsible for embryonic development and type I collagen expression.[24][25][26]
Phosphoinositide kinase FYVE mutations (formerly PIP5K3): This mutated gene, which produces proteins that help regulate endosomal function, is found in Fleck CD.[27]
COL8A2: Early-onset FECD, if inherited, is an autosomal dominant disease caused by a mutation in COL8A2.[1][28] The aberrant protein involved is alpha 2 collagen VIII.[6] Mutations in the FCD1, FCD2, FCD3, and FCD4 genes that can occur in late-onset FECD.[1][7][24][28]
Epidemiology
Fuchs endothelial corneal dystrophy is the most common among the corneal dystrophies.[6] Globally in 2012, 39% of corneal transplants were to correct FECD.[24] Even so, corneal dystrophies are rare diseases. In the United States, less than 0.01% of Americans are affected by CD.[29] Another study showed that around 530 persons out of a million in the United States have an endothelial-type CD, and 5-10 persons per million have a granular, macular or lattice-type CD.[29] Only four families have been reported to have CSCD.[4]
Demographically, CD has a variety of presentations. Both genders are equally affected by CD, although it has been shown that Fuchs CD is more common in females.[28][30] FECD is more prevalent in patients of Caucasian descent.[24] CD can be more prevalent based on global regions, such as GCD type II being predominantly European, GDLD common in Japan, Meesmann CD is found in families with German and Danish descent, and LCD type II being found in patients of Finnish descent.[4][31][32] Some dystrophies present as early as birth, such as Fleck CD, CHED, and CSCD, while others develop later on in life, typically in the second or third decade of life, such as late-onset FECD and LCD type II.[1][7][4][24]
Pathophysiology
The cornea consists of several layers. From superficial to deep, the layers are squamous epithelium, anterior basement layer (Bowman’s), a stroma comprised of keratocytes and collagen, a posterior basement membrane (Descemet’s), and an endothelial layer. The major function of the cornea is to provide a medium in which light may be refracted and focused on the retina for visual acuity.[33] The cornea is a transparent barrier between the inner constituents of the eye and the outside world and is bathed in the tear film, which creates a smooth surface for the refraction of light as well as immunologic protection.[33]
Defining corneal dystrophies depends on which layers of the cornea are affected. Alterations in proteins cause deposits within the cells of the cornea, leading to abnormal vision examination findings.[4] In fact, because of our historical limitation in understanding genetics, CD was formerly diagnosed based on the patterns of deposits found.[1] These patterns include granular, lattice, gelatinous, and macular dystrophies, among others.[1] As the stroma is the thickest portion of the cornea, and hence, the most responsible for transparency and light refraction, the accumulations of deposits often found in CD interfere with refraction, thus leading to loss of visual acuity.[33]
The squamous epithelium is highly innervated by unmyelinated nerves, which function to increase tear production and to signal the blink reflex.[33] It is nourished and oxygenated through tears since no blood vessels penetrate through to the outermost layer.[33] Patients with epithelial-type CD will be predisposed to corneal erosions, in which the patient suffers ocular pain due to the disruption and sloughing of the epithelium, especially during nighttime hypoxia.[5] Another common complication in CD is corneal haze, which is caused by inflammatory mediators and subepithelial fibrosis of Bowman’s membrane and the overlying stroma. This fibrotic change leads to loss of visual acuity.[5]
The primary function of Descemet’s membrane and the endothelial cells is to pump ions against a gradient to maintain corneal dehydration or deturgescence.[34] Gap junctions in between endothelial cells, as well as hemidesmosomes attaching to the posterior basement membrane, serve as a barrier against aqueous humor entering the drier environment of the cornea.[33] In FECD, a decrease in the number of Na/K ATPase pumps in the endothelium leads to stromal edema.[6][24][35] In addition, the proteins involved with the genetic mutations undergo a process called the unfolded protein response, which sends cells with these proteins into apoptosis, which occurs in the endothelial cells of FECD patients.[28]
Amyloidosis is a disease of abnormal protein deposition in tissues.[36] Amyloid-type protein deposition can be a systemic disease or a local manifestation, depending on the aberrant protein.[36] It is important to note that some variants of CD fall under the category of amyloidosis, ranging from systemic diseases, such as LCD type II, to localized corneal deposits, such as found in GCD, that stain appropriately for amyloid protein.[1][36]
History and Physical
Patients with CD may be asymptomatic or report blurriness of vision.[1][4] Symptomatic patients complain of eye pain, with or without light sensitivity and foreign body sensation.[1][37] These are due to corneal erosions, or the separation of the squamous epithelial layer from the anterior basement membrane.[5] Pain typically occurs in the morning, after nocturnal drying of the cornea causes the epithelium to slough and be sheared off upon eyelid opening[5] Corneal erosions are common in almost all of the epithelial-based dystrophies; therefore, all the variants should be considered in the differential diagnosis.[1][4][5] Generally, CD presents with visual alterations bilaterally. However, some dystrophies such as Posterior Polymorphous CD may present unilaterally.[1] Congenital stromal CD presents with strabismus at birth.[38]
Slit-lamp (biomicroscopy) examination findings are usually the first step in differentiating the different types of corneal dystrophies. Symptoms often group among the dystrophies based on anatomic location. An overview of typical symptoms and biomicroscopic findings are presented below, organized by anatomical classification. A more detailed description of these findings will be mentioned in other StatPearls articles on each separate dystrophy. The symptoms and findings are:
Epithelial and subepithelial dystrophies: Patients will usually suffer from recurrent corneal erosions with these dystrophies, with different degrees of severity. History and biomicroscopy findings are as follows:
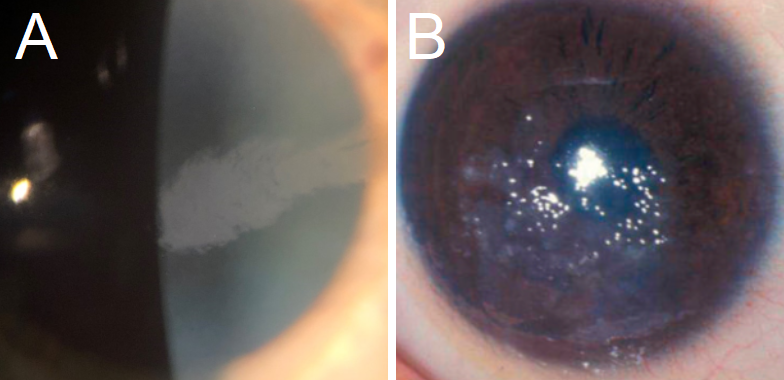
EBMCD: This CD presents with irregular, gray, map-like patches or intraepithelial (Cogan) microcysts.[1] Subepithelial pebble-like inclusions (Blebs) have been identified (see figure 1).[1] Symptoms include mild to severe corneal erosions, worse in the morning, as well as blurriness, photophobia, and diplopia.[5]
Meesmann CD: While MECD typically presents asymptomatically, or with mild symptoms of visual acuity loss and eye irritation, it can progress to painful erosions due to foreign body sensation, and potentially be severe enough to cause significant subepithelial fibrosis and vision loss.[1][19] Other common symptoms include photophobia and increased lacrimation.[32] Biomicroscopy presents with microcysts described as tiny bubbles at the level of the epithelium (see figure 1), usually in the interpalpebral area, but it has been described both centrally and peripherally.[2][4]
ERED: The recurrent erosions start in the first decade, and episodes can be severe and last for up to a week.[5] These erosions often occur at nighttime.[1] On slit-lamp exam, and patients may present with diffuse bilateral subepithelial opacifications, potentially progressing to keloid-appearing nodules, later in life.[39][40]
Subepithelial Mucinous CD: Similar to ERED, SMCD may present with subepithelial opacifications, with most of the findings in the center of the cornea.[39][40] As opposed to ERED, painful erosions tend to subside in adolescence.[1]
Lisch Epithelial CD: presents with gray whorled to band or club-shaped densities, even feather-shaped (see figure 2).[1][41] Patients may present with painless mild visual acuity loss, but many are asymptomatic until the sixth decade.[4][41]
Gelatinous Drop-Like Dystrophy: GDLD starts with a presentation similar to band keratopathy, but it progresses to mulberry-like subepithelial nodules that grow in size over time (see figure 2).[1][31] These nodules significantly alter vision. Symptoms and signs include photophobia, increased superficial vascularization with redness, and increased tear production.[4]
Epithelial-Stromal Dystrophies: Not only will patients have corneal erosions, but visual acuity loss and photophobia may occur as well, due to stromal involvement.
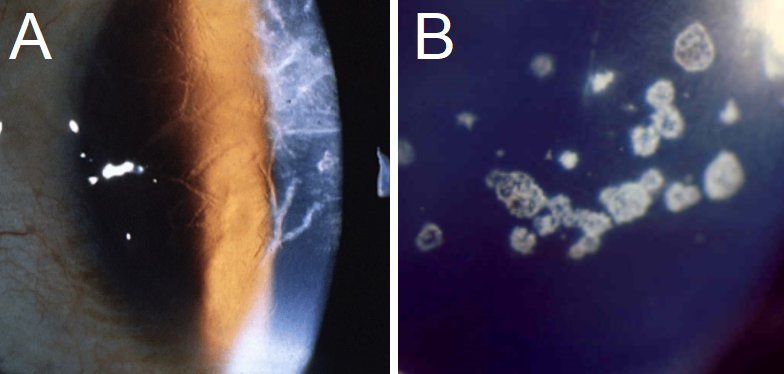
Lattice CD: On biomicroscopy, LCD presents with linear/branching opacities, forming a ropy lattice structure (see figure 3).[1] the amyloidotic ocular symptoms from LCD type II are commonly found in conjunction with Meretoja syndrome, which involves glaucoma, systemic findings such as cutis laxa (loose skin), and a signature lax, mask-like face due to facial nerve paralysis.[1][17][42] Some LCD variants appear in the 4th decade of life, presenting with similar signs and symptoms.[13]
Granular CD: If patients are symptomatic, corneal erosions starting in the first decade are likely to be seen. Visual imparity usually presents in the 5th decade of life. Biomicroscopy will present with crumb or flake-like opacities, with the spaces between the opacities clear of deposits (see figure 3).[4] Type II presents with granular and lattice-like findings on the cornea.[2]
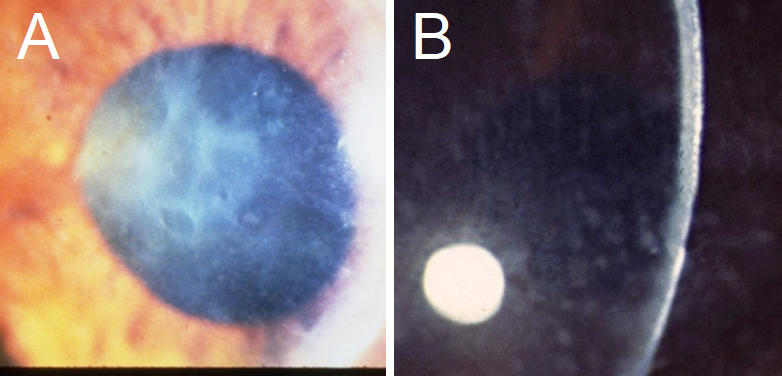
Reis-Bücklers CD: This dystrophy can present in childhood with geographic, map-like findings similar to EMBCD (see figure 4).[43] The patient will also suffer from painful erosions.[4] These will tend to fade away by the second or third decade of life, but visual imparity is often seen at this time and is associated with photophobia and corneal haze.[1][2][4][44]
Thiel-Behnke CD: The patient will present with symptoms like RBCD; however, they tend to be milder in TBCB, with visual impairment beginning later in life, and erosions starting in the first or second decade.[1][4] This dystrophy presents with honeycomb-like opacities, and the corneal surface will appear irregularly raised.[44]
Stromal Dystrophies: Corneal haze is often noted in these dystrophies, which affects visual acuity in most patients.
Macular CD: MCD presents with corneal haze and grainy, ground glass-like opacities, made of glycosaminoglycans. (see figure 4).[4] Its presentation on biomicroscopy has often been closely compared to GCD, and the hazy appearance in between opacities is indicative of MCD.[2] Typical symptoms include severe visual acuity loss and photophobia in the second to the third decade, along with mild erosions.[1] Corneal thinning is present, which additionally leads to vision impairment.[2][4] This dystrophy has been associated, rarely, with keratosis follicularis spinulosa decalvans, which causes hair loss of the scalp, eyebrows, and eyelashes, and a mutation in the CDH3 gene.[8]
Schnyder CD: Early findings by the second decade involve central crystalline subepithelial deposits with a stromal haze that progresses to a ring-like structure.[20] Arcus lipoides is often seen by the third decade.[20] Schnyder CD can present with hypercholesterolemia and other systemic lipidic symptoms like xanthelasma.[4][20] Genu valgum has been noted in some cases.[4] In addition to blurriness, advanced SCD can present with glare and decreased corneal sensitivity.[1]
Congenital Stromal CD: CSCD typically presents with symmetric corneal clouding with white flaky stromal opacities.[1] The presentation is severe enough to cause significant vision loss.[1] Some patients present with strabismus and/or open-angle glaucoma.[4][2]
Central Cloudy Dystrophy of François: CCDF presents with a lizard-like patterned gray stromal opacities, called crocodile shagreen.[1][2] There are also areas of indistinct polygonal gray zones of stroma, surrounded by normal-appearing tissue.[45] Most patients are asymptomatic.[1]
Fleck CD: This dystrophy presents with small white oval dandruff-like opacities.[27] Most patients are asymptomatic.[2]
Posterior Amorphous CD: PACD presents with sheet-like opacities, both centrally and peripherally, along the posterior stroma.[46] Onset is in very young patients, such that this disease may be considered congenital.[1] While iridocorneal adhesions and corneal thinning are noted, symptoms of glaucoma are not typical. Visual acuity is mildly affected.[4]
Pre-Descemet CD: This dystrophy presents with a condition similar to cornea farinata, which describes the accumulations as gray-white dust-like opacities made of lipids on biomicroscopy.[2] These lesions usually present later in life, and most patients are asymptomatic.[2] Pre-Descemet CD has often been found in conjunction with other disorders, such as ichthyosis.[47]
Endothelial dystrophies: Patients with these dystrophies have blurriness and visual disturbances such as haloes around light, due to corneal edema and thickness.
Fuchs Endothelial CD: FECD presents with guttae, or drop-like conglomerates, along Descemet’s membrane (see figure 5).[7] These excrescences form in areas of decellularization along the endothelial layer.[6] Signs and symptoms include corneal edema, which presents as diffuse corneal haziness that affects visual acuity.[7] Typically, blurriness is worse in the morning due to an increase in edema at nighttime.[1] Pain and photophobia are possible due to epithelial erosions caused by fibrosis and edema.[6] FECD is also evaluated using the Krachmer scale, which counts the number of guttae found on slit-lamp examination.[6] Guttae tend to start in the center of the cornea.[48] A Krachmer score of 0 means no guttae present, 1 designates 0 to 12 central guttae, 2 is greater than 12 nonconfluent guttae, 3 is 1 to 2 mm of confluent guttae, 4 is 2 to 5 mm confluent guttae, while a score of 5 signifies greater than 5 mm of confluent guttae.[6]
Congenital Hereditary CD: CHED presents with diffuse ground glass-like corneal haze and edema, along with focal gray spotting.[1][2] These cause blurry vision.[2] The autosomal recessive variant of CHED is sometimes associated with deafness.[4] A significant increase in corneal thickness is noted in this disease.[4] Nystagmus is present.[1]
Posterior Polymorphous CD: PPCD can present with a variety of asymmetric or unilateral biomicroscopic findings, including vesicles, breaking bands, or gray haze (see figure 5).[1][49] The visual disturbances and pain associated with glaucoma can also present in PPCD, as it tends to form peripheral anterior synechiae (iris-angle adhesion).[2][25] Keratoconus is often seen in patients, leading to visual acuity loss, along with corneal edema and photophobia.[1][2] On light microscopy, it is noted that the endothelial layer changes to stratified squamous epithelium.[4]
X-linked Endothelial CD: XECD presents with diffuse milky opacities with band keratopathy as well as moon crater-like endothelial cells.[50] It is of note that moon crater change in endothelial cells is only seen in female carriers.[1] In addition, affected males have blurred vision and nystagmus, while females are asymptomatic.[1][4]
Evaluation
Diagnosis begins with a full eye examination, with emphasis on biomicroscopy, which was the original technique to observe the physical findings on the cornea.[1] Light microscopy of histologic specimens can help identify subtypes of CD.[1] EBMCD and Meesmann CD display a thickened Bowman’s Layer.[1][2] GCD type 1 presents with granular, snowflake-like hyaline deposits, while LCD presents with amyloid-like inclusions.[2] GCD type 2, or Avellino-type, can present with both hyaline and amyloid-like deposits histologically.[2][4] In FECD, there is a thickened Descemet’s membrane with guttae expressed as hyaline outgrowths from the membrane.[1] Reis-Behnke CD and GCD are positive for rod or trapezoidal-like inclusions found in the stroma.[2][4] Schnyder CD finds cholesterol deposits in the stroma.[2][20]
Staining of histologic slides is crucial to diagnose some forms of CD, as it may lead to the type of substance deposited in the cornea. Many dystrophies that include a mutation in TGFBI, like LCD and GCD, will stain positive for Congo red, as these dystrophies produce an amyloid-like disease.[2][15] Other amyloid positive dystrophies, such as GCD, also stain positive with Masson Trichrome and show eosinophilic deposits on H&E.[1][4] Trichrome staining is also helpful for distinguishing the subepithelial mucoid material found in SMCD.[4] Periodic acid-Schiff (PAS), which stains polysaccharides and glycogen, can be positive in Meesmann CD, as well as highlight the intraepithelial cysts found in the disease.[1][2] Alcian-blue, which stains for acid mucins, will stain positive in Fleck CD, MCD, ERED, and Subepithelial Mucinous CD.[1][4] In addition, colloidal iron will be positive in MCD, due to the accumulation of glycosaminoglycans in all layers of the cornea.[22] Drop-like gelatinous CD stains positive for lactoferrin antibodies, even though the deposits are amyloid in nature.[1][4] Posterior Polymorphous CD will stain positive for cytokeratin, which stains proteins involved in the cytoskeleton of a cell.[1]
Transmission electron microscopy (TEM) is often involved in academic investigations of CD, but it can also be used to confirm the diagnosis in some dystrophies.[1][4] Certain characteristics found on TEM distinguish one variant of CD from another; for example, in Meesmann CD, collections of fibrogranular material, as well as mutated cytokeratin, known as “peculiar substance,” can be found in the cytoplasm.[1][4] TBCD presents with curly subepithelial fibrils.[4] Findings in MCD include intracytoplasmic vacuoles with fine fibrillogranular material.[4]
Treatment / Management
In patients who are asymptomatic or mild cases of CD, regular follow-up with the clinician is recommended for any typical ophthalmologic changes, as well as the possibility for any disease progression.[4] Regular follow-up appointments for the slow-progressing variants are more crucial for proper intervention, with typical conservative management including corrective lenses.[4]
Treatment for symptomatic dystrophies can be organized by anatomic location, as symptoms tend to become similar among dystrophies of the same classification. Elaboration on management for each type of dystrophy will be discussed in other StatPearls articles.
Epithelial dystrophies: The most common symptoms seen are corneal erosions, especially in the morning, due to the increase of epithelial sloughing from nocturnal corneal drying.[5][51] Symptomatic management for recurrent corneal erosions starts with artificial tear administration to lessen friction caused by blinking, therefore preventing epithelial sloughing.[5] Hypertonic nighttime lubricants can help reduce friction as well as decrease corneal edema in the intracellular space.[5][51] NSAIDs can help reduce pain, as well as bandage contact lenses.[5][37] In patients with more severe erosions, superficial keratectomy, anterior stromal puncture, and phototherapeutic keratectomy (PTK) are treatment options that may help alleviate symptoms.[5][52]
Stromal dystrophies: CD involving the stroma may most likely lead to visual acuity loss.[4] As some dystrophies involve both the epithelium and stroma, they are often associated with corneal erosions as well.[4] In addition to conservative treatment, as mentioned in the epithelial section before, invasive measures may be needed to resolve symptoms. As opposed to PTK, automated lamellar keratoplasty (ALK) uses a microkeratome to cut deeper in the corneal surface, roughly 100 to 300 microns, to remove aberrant cells involved in the stroma.[53] In patients with dystrophy involvement down to the posterior basement membrane (Descemet’s), deep automated lamellar keratoplasty (DALK) may be performed to remove more corneal tissue.[54]
Endothelial Dystrophies: With the advent of Descemet’s stripping endothelial keratoplasty (DSEK) and Descemet’s membrane endothelial keratoplasty (DMEK), the need for penetrating keratoplasty has diminished due to faster recovery, better post-surgical visual acuity, and decreased graft rejection, because there is no impact on the anterior face of the ocular surface with these procedures.[7][28]
Other techniques that are used presently or may be implemented in the future consist of using gene or enzyme therapy, mesenchymal stem cells to replace dystrophic cells, and lipid-based therapies.[22][55] Rho-kinase inhibitors could be used to help decrease intraocular pressure in dystrophies causing glaucoma symptoms, as well as increase endothelial cell adhesion and replication in endothelial dystrophies such as FECD.[56] As CD is predominantly a genetic disease, genetic counseling is recommended to those planning to have children.[4]
Differential Diagnosis
It is often the case that corneal dystrophies must be differentiated among each other, as clinical presentations may often overlap. While biomicroscopy findings may identify different types of CD, these findings may be similar among dystrophies. Genetic testing may aid in the differentiation among dystrophies of similar presentations.
The presenting signs and symptoms of CD may be due to other ocular diseases. Recurrent corneal erosions may be caused by non-genetic etiologies.[5] Corneal degeneration may be due to the aging process or trauma.[5] Furthermore, the elderly can present with endothelial guttae and basement membrane degeneration without having an etiology of corneal dystrophy.[57] Other etiologies include infectious keratitis, either bacterial or commonly from Herpes, and neurotrophic keratitis, where degeneration of the cornea happens due to trigeminal nerve damage and desensitization.[51][58][59] Band keratopathy, or the deposition of calcium in the cornea, can come from chronic uveitis, systemic diseases of hypercalcemia, and sarcoidosis, among other diseases.[58] Salzmann nodular degeneration is an idiopathic process where bilateral blue-white nodules of hyaline are deposited underneath Bowman’s membrane.[58] The crocodile shagreen seen in central cloudy dystrophy is typically an idiopathic finding in the elderly. So dystrophy should be included in the differential if these findings are present in children and adolescents.
A patient with corneal erosions could also be affected with Herpes keratitis, infectious keratitis, conjunctival foreign bodies, dry eyes secondary to autoimmune disease, among other pathophysiological processes.[5] As mentioned before, ocular pain and visual disturbances found in glaucoma can present similarly in some types of CD2 or even be in conjunction with CD, as is the case with congenital stromal CD.[4] Sometimes, the differential could include other types of corneal dystrophy, as they tend to present with similar symptoms. For example, thorough genetic testing can help differentiate between Meesmann CD and Lisch epithelial CD, as well as all the dystrophies that cause recurrent corneal erosions.[4] GCD can present with similar lesions found in monoclonal gammopathies.[4] SCD can be found with systemic signs of hyperlipidemia, which are similarly found in inheritable diseases such as lecithin: cholesterol acyltransferase (LCAT) disease.[4] Diseases that affect basement membranes, such as Alport syndrome and Usher syndrome, could potentially present with ocular symptoms like CD.[60][61]
Prognosis
The prognosis of CD depends mainly on the subtype of dystrophy. However, most CD presents with progressive loss of visual acuity.[4] Posterior Amorphous CD is virtually asymptomatic, only presenting with mild visual refractive error.[1][4] Some dystrophies, like posterior polymorphous CD, Meesmann CD, and Fleck CD, present early in life but tend to progress slowly, requiring little treatment in some cases.[2][4] Others, like FECD, will progress more rapidly, enough to warrant surgical intervention by the sixth decade of life.[1] Also, MCD, GDLD, and congenital stromal CD can present severely early in onset, threatening severe vision loss in patients who do not receive keratoplasty.[2]
There is a chance that recurrence of the dystrophy can happen post-graft transplantation, requiring retreatment. It is more likely to occur in stromal dystrophies.[4] For example, it is reported that LCD recurrence after keratoplasty at nine years is 60%, and GCD may reoccur on an average of 2-3 years post-surgery.[4][53][54] Recurrence has been reported in other types of dystrophies as well, albeit rarely.[62][63] It should be stated that retreatment is not always due to the recurrence of the disease. In patients with FECD, repeat corneal grafting is most likely due to donor corneal detachment, failure, or endothelial cell-loss, rather than recurrence of the dystrophy.[7]
Complications
Complications from CD can arise from untreated disease, and there are always risks of complications involved with surgical intervention. If not treated, patients with severe forms of CD may develop progressively worse vision, to the point of severe visual impairment.[1][22] Corneal erosions are painful and can lead to secondary keratitis due to excessive rubbing by the patient.[64] As treatment primarily involves removing the affected corneal tissue and transplanting donor tissue, the risk of infection, graft rejection, and treatment failure are present. It is reported that graft rejection can happen in up to 35% of corneal dystrophy patients within one year.[65]
Deterrence and Patient Education
It is important to let patients understand the limitations of surgical intervention. Many dystrophies can reoccur in a few years, despite corneal transplant.[21] For CD that progresses slowly, non-invasive treatment with spectacles and contact lenses is recommended.[21]
Patients should be educated on post-surgical safety measures to promote optimal outcomes. Bandage contact lenses may aid in preventing loose sutures and displacement of the graft.[64]
The application of artificial tears in the morning may help alleviate corneal erosion pain.
Pearls and Other Issues
Corneal dystrophies are a collection of inheritable diseases leading to cellular dysfunction of the cornea.
Patients with corneal dystrophy range from being asymptomatic to displaying symptoms such as painful corneal erosions, photophobia, and visual acuity loss, and can be associated with other systemic symptoms based on the genetic abnormality.
Physical exam findings, especially biomicroscopy, histologic exam, and genetic testing, are vital to the accurate diagnosis of corneal dystrophies.
Treatment can vary from conservative measures to corneal transplantation and is dependent on disease onset and progression.
Enhancing Healthcare Team Outcomes
Management of corneal dystrophy is a combined effort by many medical professionals. Despite the rarity of these diseases, optometrists and ophthalmologists performing routine screening exams on patients should always be aware of the slit-lamp findings that are pathognomonic of CD. A clinician, as well as medical assistants, technicians, and nurses, should be aware of symptoms and signs of CD and be thorough patient history gathering, especially family history, to increase the chance of recognizing potential dystrophy. Proper communication among team members of these clinical findings is crucial to appropriate diagnosis through a physical exam and laboratory testing. Over-testing should be avoided to limit the cost to the patient. Finally, proper communication and education should be given to the patient regarding diagnosis and treatment, follow-up, and potential surgical intervention. It is noteworthy that the ICD-10 codes for CD are divided into six categories: endothelial, epithelial (Juvenile’s), granular, macular, unspecified, and other hereditary corneal dystrophies.