Continuing Education Activity
Osteosarcoma is a malignant osseous neoplasm. It is the most common primary bone cancer of childhood. Less frequently, it occurs in adults where it represents secondary malignant degeneration of primary bone pathology. This activity reviews the clinical presentation, evaluation, and management of osteosarcoma, and highlights the role of an interprofessional healthcare team in treating patients with this condition.
Objectives:
- Summarize the pathophysiology of primary and secondary forms of osteosarcoma.
- Identify the typical imaging findings associated with osteosarcoma.
- Review the treatment considerations for patients with osteosarcoma.
- Outline the importance of collaboration and coordination among members of the interprofessional team in managing patients with osteosarcoma.
Introduction
Osteosarcoma is the most common primary pediatric bone malignancy, derived from primitive bone-forming (osteoid producing) mesenchymal cells. It occurs in primary (no underlying bone pathology) and secondary forms (underlying pathology which has undergone malignant degeneration/conversion), accounting for approximately 20% of all primary bone tumors. Osteosarcoma is highly heterogeneous in its manifestation, which permits division into several subtypes according to the degree of differentiation, location within the bone, and histological variation. These subtypes vary in imaging appearance, demographics, and biological behavior. With the ceaseless work of numerous medical, surgical, and scientific professionals, treatment options and survivability have vastly improved in recent years.[1]
Etiology
Although the quality of life of patients affected by osteosarcoma has significantly improved over the last few decades, its etiology remains obscure. Studies aiming to determine the causes of osteosarcoma have classically focused on multiple factors, including genetics, epidemiology, and the environment.[2]
The highly complex karyotypes typical of osteosarcoma tumor cytology have created challenges regarding the thorough characterization of recurrent chromosomal mutations.[2] However, research has identified several genetic aberrations in cases of primary osteosarcoma:
- Hereditary Retinoblastoma: An autosomal dominant condition caused by germline mutations in the RB1 gene, causes bilateral retinoblastoma at an average presenting age of one year. Retinoblastoma characteristically presents as an absence of the "red reflex" in the eye or eyes of the affected child. This disorder imparts an increased risk of osteosarcoma later in life.[2]
- Li-Fraumeni Syndrome: An autosomal dominant disorder due to mutations in the p53 tumor suppressor gene, has been found in up to 3% of children with osteosarcoma. Patients with this disorder are also at a high risk of developing several additional types of cancer at a very early age.[2]
- Rothmund-Thompson Syndrome: An autosomal recessive syndrome due to a mutation in the RECQL4 gene, conveys a predisposition to osteosarcoma as well as a characteristic infantile rash, dysplastic osseous structures, alopecia, premature cataracts, and chronic gastrointestinal distress.[2]
- Bloom Syndrome: An autosomal recessive disorder caused by mutations in the BLM gene, a gene responsible for maintaining DNA stability during replication. In addition to a predisposition to osteosarcoma and other cancers, these patients may also present with UV-induced rashes, short stature, and sparse subcutaneous fat.[2]
- Werner Syndrome: An autosomal recessive disorder, also known as adult progeria, is characterized by premature aging, bilateral cataracts, osteoporosis, short stature, scleroderma-like skin changes, and a predilection for osteosarcoma. A faulty WRN gene is responsible.[2]
Research has identified associations with secondary osteosarcoma in patients with Paget disease, electrical burns, trauma, exposure to beryllium, exposure to alkylating agents, FBJ virus, osteochondromatosis, enchondromatosis, fibrous dysplasia, orthopedic prosthetics as well as bone infarction and infection. Additionally, osteosarcoma reportedly correlates with exposure to ionizing radiation, radium, and archaic contrast agents such as thorotrast.[3][4]
Epidemiology
Osteosarcoma has a bimodal age distribution. The initial peak is in the 10 to 14 year age group, corresponding to the pubertal growth spurt. This group represents the vast majority of primary osteosarcomas. In the 0 to 14 year age range, the incidence rate of osteosarcoma in all races and genders is four cases per year per million people (3.5 to 4.6, 95% confidence interval). This number rises to five cases per year per million people (4.6 to 5.6, 95% confidence interval) for the range 0 to 19 year age range. The next observable peak is in adults older than 65, when the appearance of osteosarcoma is more likely to represent secondary cancer resulting from malignant degeneration of Paget disease, sites of bone infarction, etc. The patient’s age has been found to correlate with survival; the poorest survival is among older individuals. Death rates for osteosarcoma have steadily declined by approximately 1.3% per year. The 5-year overall survival rate is about 68%, regardless of sex.[5]
Osteosarcoma accounts for approximately 2.4% of pediatric cancers, making it the eighth most common childhood malignancy. Leukemia is the most common (30%), followed by malignancies of the central nervous system (22.3%), neuroblastoma (7.3%), Wilms tumor (5.6%), non-Hodgkin lymphoma (4.5%), rhabdomyosarcoma (3.1%), and retinoblastoma (2.8%).[5]
Blacks are the ethnic group most likely to be affected by osteosarcoma, with an incidence rate of 6.8 cases per year per million people. Hispanics are a close second with an incidence rate of 6.5 cases per year per million people. White race individuals experience this malignancy at a rate of 4.6 cases per year per million people.[5]
The incidence of osteosarcoma has historically been reported as higher in males than in females, with an incidence rate of 5.4 cases per year per million males and 4 cases per year per million females, respectively.[5]
Pathophysiology
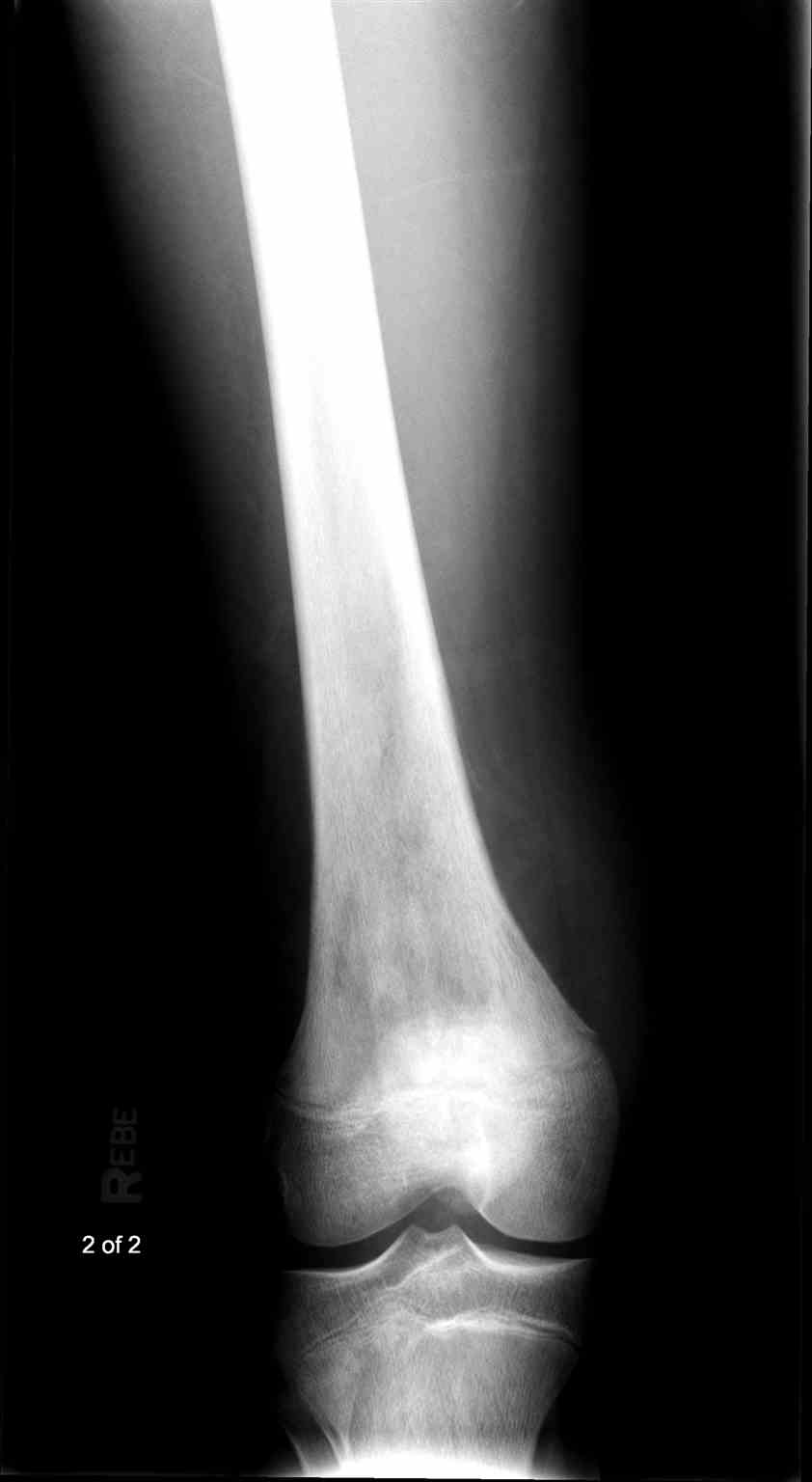
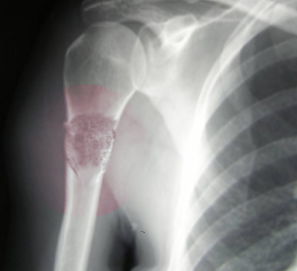
Osteosarcoma frequently occurs near the metaphysis of the long bones of the appendicular skeleton. The most common locations include the femur (42%, with 75% of tumors in the distal portion of the bone), the tibia (19%, with 80% of tumors in the proximal portion of the bone), and the humerus (10%, with 90% of tumors in the proximal portion of the bone). Other potential sites include the skull or jaw (8%) and the pelvis (8%).[5] As mentioned previously, osteosarcoma can subdivide into primary and secondary forms:
- Primary tumors usually occur in the metaphysis of long bones and have a marked predilection for the knee, with nearly 60% occurring at this location. Children and adolescents account for the vast majority of those affected by this condition.[5]
- Secondary tumors have a much wider distribution, reflecting the varied nature of their underlying predisposing condition. They almost always occur in the adult population. Notably, they feature a higher incidence in flat bones, the pelvis in particular (a common location of Paget disease).[5]
Histopathology
Osteosarcoma is grossly divided into various subtypes based on its location within the bone then further subdivided by grade.
- Central (Intramedullary):[6]
- High-Grade Central
- Conventional – “classic” appearance; cells are spindle-like to polyhedral in shape; nuclei are variable in appearance, and cells undergoing mitosis are readily identifiable. Matrix production by the tumor cells may be osseous (“osteoblastic”), cartilaginous (“chondroblastic”), or fibrous (“fibroblastic”) but a combination of the three often presents. Osteoid matrix must be identified somewhere in the lesion, even if only in a minuscule amount. In the case of osteoblastic osteosarcoma, there may be excessive osteoid matrix production such that the tumor is described as “sclerosing” in appearance.
- Osteoblastoma-like – histologically resembles osteoblastoma but features more cellular atypia and local aggressiveness, and osteoid matrix production
- Chondroblastoma-like – histological resembles chondroblastoma but features more cellular atypia, local aggressiveness, and osteoid matrix production
- Chondromyxoid fibroma-like – histological resembles chondromyxoid fibroma but features more cellular atypia, local aggressiveness, and osteoid matrix production
- Malignant fibrous histiocytoma-like – histologically resembles malignant fibrous histiocytoma but features more cellular atypia, local aggressiveness, and osteoid matrix production
- Epithelioid – features cells that are so poorly differentiated that it may be hard to distinguish histologically whether the lesion is a sarcoma (connective tissue origin) or a carcinoma (epithelial origin)
- Giant cell – features benign multinucleated osteoclast-like giant cells
- Clear cell – features numerous cells with clear or ground-glass cytoplasm and vacuoles
- Telangiectatic – comprised of numerous blood-filled sinusoids such that the tumor mimics the histology of an aneurysmal bone cyst; the presence of pleomorphic/atypical nuclei will distinguish the malignancy.
- Small Cell – considered to be a histological combination of Ewing sarcoma and osteosarcoma, features numerous small round cells. A minuscule volume of an osteoid matrix will distinguish this as a variant of osteosarcoma.
- Low-Grade Central
- Fibrous dysplasia–like – features a large volume of osseous matrix embedded in a small amount of fibrous stroma.
- Desmoplastic fibroma–like – features a very small volume of osseous matrix embedded in a large amount of fibrous stroma.
- Surface (Periosteal/Cortical):[6]
- Low-Grade Surface
- Parosteal – found on the outer periosteal surface of the bone, features ribbons of osseous trabeculae oriented in parallel, primarily composed of a chondroid matrix with only a minuscule amount of osteoid matrix.
- Intermediate-Grade Surface
- Periosteal – found between cortex and the inner periosteal surface of the bone, features ribbons of osseous trabeculae oriented in parallel, primarily composed of a chondroid matrix with only a minuscule amount of osteoid matrix, more nuclear atypia than the parosteal variant.
- High-Grade Surface
- High-Grade Surface – histological identical to high grade/conventional/central variant, varies only in location (being confined to the surface of the bone) which is thought to represent dedifferentiated parosteal osteosarcoma.
- Extraskeletal:[6]
- Low-Grade-Histologically identical to low-grade surface/parosteal variant and low-grade central variant varies only in geography, potentially appearing at any extraskeletal location in the body, including the soft tissues of the thigh, buttocks, upper extremities, or retroperitoneum.
- High-Grade-Histological identical to high grade/conventional/central variant differs only in geography, being found at any extraskeletal location in the body.
History and Physical
Symptoms of osteosarcoma may be present for a significant amount of time, sometimes weeks to months, before patients seek evaluation. Most commonly, the presenting symptom is bone pain, particularly with activity. Parents are often concerned that their child has incurred a sprain, arthritis, or growing pains. There may or may not be a reported history of traumatic musculoskeletal injury.[7]
Pathologic fractures are not a common mainstay of osteosarcoma, except for the telangiectatic type of osteosarcoma, which is associated with pathologic fractures. The resulting pain may manifest as a limp. A swelling or lump may or may not be reported, depending on tumor size and location. Systemic symptoms, such as those seen in lymphoma (fever, night sweats, etc.), are rare.[7]
Respiratory symptoms are rare and, when present, indicate extensive lung involvement. Additional symptoms are unusual because metastases to other sites are extremely rare.[7]
Physical examination findings are typically focused around the location of the primary tumor and may include:[7]
- A palpable mass may be tender and warm with or without an overlying pulsation or bruit, though these signs are nonspecific
- Joint involvement with decreased range of motion
- Local or regional lymphadenopathy (unusual)
- Respiratory findings with metastatic forms
Evaluation
National Comprehensive Cancer Network's 2020 Guidelines for Initial Evaluation of Osteosarcoma (Version 1.2020)
- Clinical History and Physical Exam (discussed above)
- Laboratory Analysis of Lactate Dehydrogenase (LDH) and Alkaline Phosphatase (ALP) Levels: Biochemical markers such as serum alkaline phosphatase (ALP) and lactate dehydrogenases (LDH) are assessed in the initial workup because they provide evidence for diagnosis and prognosis. ALP levels will be high due to the increased osteoblastic activity associated with osteosarcoma. Extremely high levels have been linked to heavy tumor burden and are generally considered a poor prognostic indicator. It is important to evaluate the levels of the biomarkers later in the treatment process as well, as levels may decrease with success therapy or rise with residual disease or recurrence.[8]
- Diagnostic Imaging of Primary Tumor Site
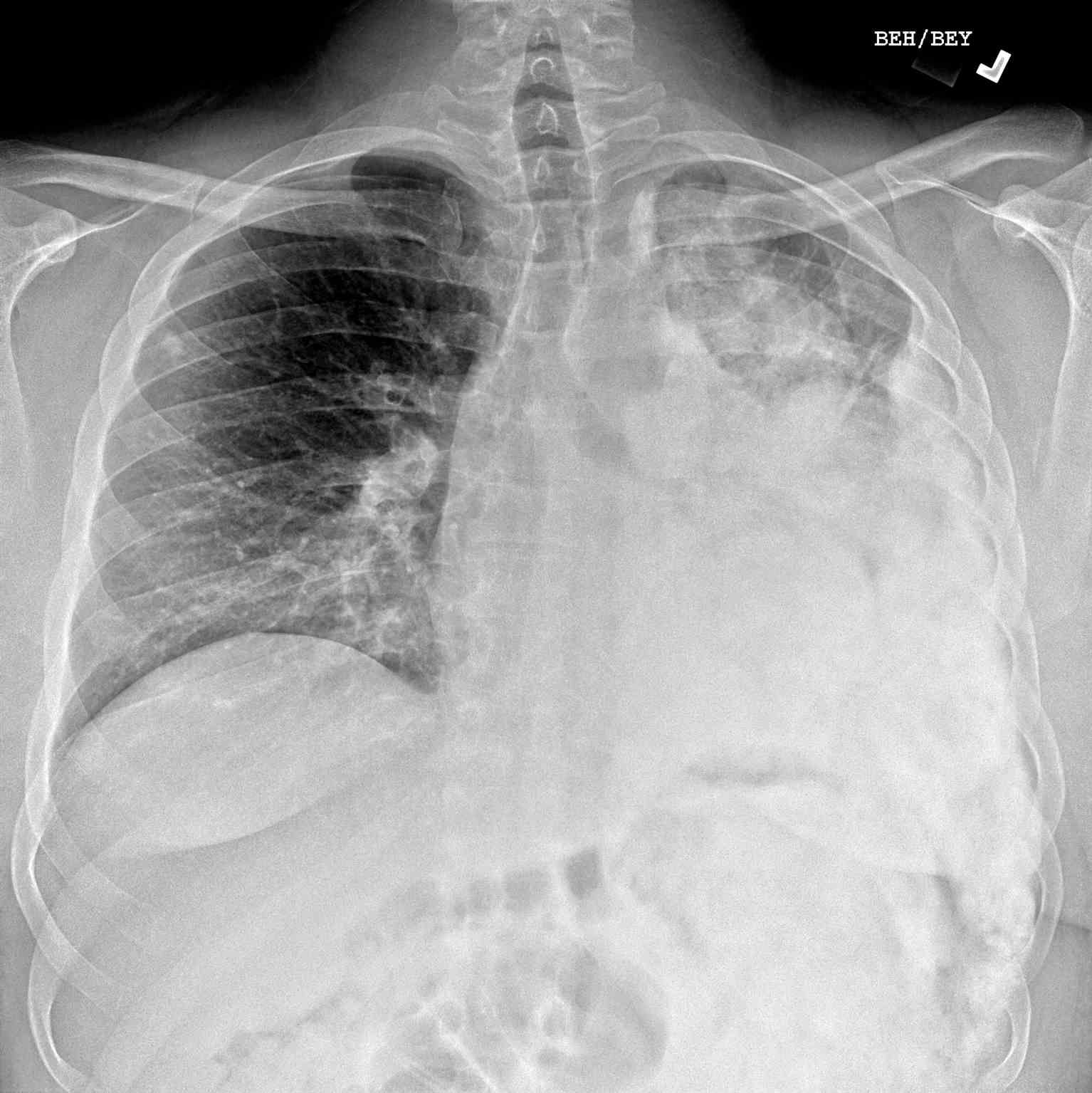
- Radiographs - although MRI is the gold standard for diagnostic imaging of osteosarcoma, radiographs are generally the first study obtained when a potential bone mass is identified on the physical exam.[8] A conventional radiograph of osteosarcoma may demonstrate- medullary and cortical bone destruction, permeative or moth-eaten cortex, "Sunburst" configuration (due to aggressive periostitis), "Codman triangle" configuration (due to elevation of the periosteum away from the bone), ill-defined "fluffy" or "cloud-like" osseous lesion, soft-tissue mass, calcification of osteoid matrix produced by the tumor.[8]
- Magnetic Resonance Imaging - after identifying a suspicious lesion on a radiograph, MRI may be necessary for further characterization. MRI is an indispensable tool for defining the extent of a tumor inside and outside the bone. The entirety of the involved bone, as well as one joint above and one joint below the tumor, should be included in the study so that “skip” lesions are not missed. MRI can accurately and precisely delineate the degree of tumor in the adjacent soft tissues, joint involvement, whether or not the tumor crosses the physis, proximity to the nearest neurovascular bundle. Nearly every aspect of treatment is assessable with MRI, from pre-surgical assessment for limb-sparing resection to the degree of chemotherapy response in the form of tumor necrosis, shrinkage, and improved capsulation.[8] Traditional sequences acquired in MRI of osteosarcoma may demonstrate the following:[8]
- T1 Weighted Images
- Non-ossified soft tissue component: intermediate signal intensity
- Osteoid components: low signal intensity
- Peritumoral edema: intermediate signal intensity
- Scattered foci of hemorrhage: variable signal intensity based on chronicity
- T2 Weighted Images
- Non-ossified soft tissue component: high signal intensity
- Osteoid components: low signal intensity
- Peritumoral edema: high signal intensity
- Computed Tomography - the role of CT is primarily to assist with biopsy planning and disease staging. CT may not significantly contribute to direct assessment of the tumor after radiography and MRI unless the osseous lesion in question is predominantly lytic. In the case of lytic lesions, small amounts of mineralized material may be unobservable on both plain film and MRI. CT of the chest, however, is the modality of choice for the evaluation of metastasis.[8]
- Nuclear Imaging
- Positron Emission Tomography – PET is a nuclear medicine imaging modality that detects highly metabolic lesions. It is an essential tool that is useful for determining tumor extent and searching for subtle lesions after identifying a suspicious mass on initial diagnostic imaging. Later in the treatment process, PET is valuable for the detection of recurrence.[8]
- Radionuclide Bone Scan - Technetium 99 methylenediphosphonate (Tc99 MDP) bone scan is an effective and readily available imaging modality for detecting bony metastasis. It is a less expensive but less specific alternative to PET imaging.[8]
- Follow up MRI or CT (both with contrast) of sites of metastasis identified on PET or bone scan
- Fertility Consultation may be a consideration (chemotherapy and radiation therapy may affect fertility).
- Biopsy of Osteosarcoma
After the physical exam, laboratory analysis, and diagnostic imaging confirm the presence of a lesion consistent with osteosarcoma, a biopsy is necessary. The final surgical procedure must include resection of the biopsy tract, which should be tattooed for easy identification, to avoid recurrence due to potential seeding of this tract with cancer cells. Ideally, the surgeon who undertakes the biopsy should be the same individual who completes the resection, so they are familiar with the path and extent of the biopsy. An open approach to biopsy was previously considered to be the best option owing to a high rate of accuracy. In recent years, however, research has determined that an open approach correlates with an increased risk of complications such as infection, improper wound healing, and seeding of the site by tumor cells, as previously discussed. As such, core biopsy has replaced the traditional open approach, particularly because of the reduced risk of contamination of the surgical bed with tumor cells but also due to lower cost and decreased recovery time. It is especially crucial for patients with perceived potential for limb-sparing procedures in which as much local tissue should be spared as safely possible. Core needle biopsy is achieved via a single deep stab with a needle through a trocar, which traverses a single tissue plane in a location that will be included in the final resection. Multiple cores are necessary from the representative region of the mass - the soft tissue portion in the periphery of the lesion. The necrotic central region will yield little viable tissue, and the “Codman triangle” region will yield only reactive bone. Importantly, recent studies have shown that fine-needle aspiration is not an efficacious approach to biopsy because it does not yield an adequate tissue sample for an accurate diagnosis. Following the biopsy, tissue samples should be analyzed by pathologists in fresh or frozen format for definitive diagnosis, grading, and histological subtyping, all of which will affect medical and surgical treatment strategy.[9]
Treatment / Management
National Comprehensive Cancer Network's 2020 Guidelines for Management of Osteosarcoma (Version 1.2020)
- OSTEO-1 (Low-Grade Osteosarcoma, No Metastasis)
- Intramedullary and surface
- Wide excision alone (no neoadjuvant chemotherapy)
- If postsurgical pathology demonstrates low-grade features, then no adjuvant chemotherapy
- If postsurgical pathology demonstrates high-grade features, consider adjuvant chemotherapy
- Periosteal
- Neoadjuvant chemotherapy then perform a wide excision
- If postsurgical pathology demonstrates is consistent with biopsy (low grade features only) then no adjuvant chemotherapy
- If postsurgical pathology demonstrates high-grade features, consider adjuvant chemotherapy
- OSTEO-2 (High-Grade Intramedullary or Surface Osteosarcoma, No Metastasis)
- Neoadjuvant chemotherapy then restage the lesion
- If restaging suggests the lesion is resectable, then perform a wide excision
- Positive margins
- If there was a good response to preoperative neoadjuvant chemotherapy (less than10% viable tumor on postsurgical pathology), then continue the same neoadjuvant chemotherapy regimen and consider additional surgical resection +/- radiation therapy
- If there was an inadequate response to preoperative neoadjuvant chemotherapy (greater than 10% viable tumor on postsurgical pathology), then continue the same neoadjuvant chemotherapy regimen or consider a new regimen and consider additional surgical resection +/- radiation therapy
- Negative margins
- If there was a good response to preoperative neoadjuvant chemotherapy (less than 10% viable tumor on postsurgical pathology), then continue the same neoadjuvant chemotherapy regimen. No further resection is required.
- If there was an inadequate response to preoperative neoadjuvant chemotherapy (greater than 10% viable tumor on postsurgical pathology), then continue the same neoadjuvant chemotherapy regimen or consider a new regimen. No further resection is required.
- If restaging suggests the lesion is unresectable, then continue chemotherapy and consider radiation therapy.
- OSTEO-3 (Any Grade With Metastasis at Presentation)
- If metastases are resectable (pulmonary, visceral, or skeletal), then perform metastasectomy and follow OSTEO-2 guidelines.
- If metastases are unresectable, then consider chemotherapy and radiation therapy, after which the primary site requires reassessment for local control.
- OSTEO-4 (Follow-up & Surveillance)
- Surveillance schedule
- Every three months for post-op years 1 and 2
- Every four months in post-op year 3
- Every six months in post-op years 4 and 5
- Yearly for post-op years six and beyond
- Surveillance visit should include
- Physical exam with assessment of function
- Imaging of post-op site and chest
- Consider PET/CT or bone scan
- CBC +/- additional laboratory tests as clinically indicated (e.g., alkaline phosphatase levels)
- If a relapse is detected, the following are the guidelines to follow:
- Chemotherapy +/- resection (if possible)
- Response to these treatments should have an evaluation via:
- Radiographs of the original tumor site
- CT or MRI (both with contrast) of the site of relapse
- CT of the chest to assess for pulmonary lesions
- Good response to treatment:
- Surveillance (restart OSTEO-4 guidelines)
- Poor response/progression of the disease:
- Resection (if possible)
- Clinical trial
- Palliative radiation
- Best supportive care
Extraskeletal Osteosarcoma
- Follow the National Comprehensive Cancer Network's guidelines for the treatment of soft tissue sarcoma.
Differential Diagnosis
While assessing a patient with osteosarcoma following differentials should be considered:
- Other types of tumor (both malignant and benign):[10]
- Osseous metastasis from other types of malignancies
- Ewing sarcoma
- Malignant fibrous histiocytoma
- Fibrosarcoma
- Giant cell tumors
- Lymphoma
- Osteoblastoma
- Cortical desmoid
- Non-tumor bone lesions:[10]
- Osteomyelitis
- Aneurysmal bone cyst
- Fibrous dysplasia
Surgical Oncology
The goal of surgical management of osteosarcoma is complete excision of the lesion, which typically takes place via resection with wide margins. Two main approaches exist to accomplish this objective: limb salvage and amputation.[11]
Limb Salvage
- The vast majority of patients (about 85 to 90%) with osteosarcoma undergo limb salvage. Limb salvage involves the removal of a tumor from a limb without the removal of the entire limb itself. This process occurs in two main steps: initial resection and subsequent reconstruction. Resection is essential for the elimination of the disease. As previously mentioned, the surgical excision of the mass should also include the biopsy site/tract, with a minimum margin of 2 cm, to avoid recurrence of disease from tumor cells, which may have escaped during tissue sampling. Radiological imaging performed during the initial evaluation should be reviewed preoperatively to determine the volume of bone that requires removal. Ideally, the mass and reactive zone surrounding it should not be disturbed, such that the entirety of dissection occurs through normal healthy tissues, which typically includes an additional 6 to 7 cm of adjacent normal bone. Computer-generated anatomical reconstructions are very useful for this purpose, particularly in tumors of the flat bone of the pelvis and sacrum, where excessive resection of bone can create postoperative problems with structural stability.[9][11]
- Because osteosarcoma is the most common primary osseous malignancy in the pediatric population, surgery presents a unique set of challenges. To achieve clear margins, excision may necessitate physeal resection, which can lead to growth disturbances as the child matures. In the past, a tumor that traversed the growth plate was considered to be an indication for amputation because there was no available means to restore function. With the advent of options that “grow” or expand with the patient (discussed below), masses that cross the growth plate are no longer considered a contraindication to limb salvage.[9][11]
- Another difficulty that surgeons encounter in the quest for limb salvage is a mass that encompasses a joint; this is a common challenge due to the predilection of osteosarcoma for the knee. However, a combination of resection and tissue regeneration (discussed below) have helped to over this obstacle such that joint involvement is no longer considered a contraindication to limb salvage.[9][11]
- After resection, reconstruction can begin. The purpose of reconstruction is the restoration of function to the affected limb. In the case of non-weight-bearing bones like the fibula or clavicle, reconstruction is unnecessary because the excision of these structures does not impart functional deficit. Reconstruction of weight-bearing bones, as one might imagine, is an arduous task. One of the greatest challenges for surgeons is a recapitulation of large swaths of missing bone. In recent years, several options have become available. These options fall into three main classes: allograft/autograft bone reconstruction, metallic (endoprosthesis) reconstruction, and tissue regeneration reconstruction.[9][11]
- Allograft/Autograft Bone Reconstruction
- Allograft bone replacement utilizes bones collected in the postmortem period from organ donors. As with organ donation, potential donors undergo screening for communicable diseases. Once surgically grafted into the osteosarcoma patient, the native bone will grow into the allograft bone and heal. Rejection is rare because very few donor cells remain within the donated bone, and the bone itself is a relatively inert material. As one might imagine, the most serious complication that may arise with allograft reconstruction is the failure of fusion between patient bone and allograft bone. Infection and fracture are also important complications that require internal fixation or removal, respectively. A hybrid reconstructive device is available in the form of an allograft prosthetic composite (APC), which combines an allograft bone fragment with a metallic prosthesis. APC arthroplasty is useful for the reconstruction of weight-bearing joints such as the knee or hip. This device combines the easier reinsertion of a biological graft with the instant weight-bearing ability of a prosthetic.[9][11]
- In centers without access to a donor bone bank, resected malignant bone can be irradiated (or less frequently, pasteurized or treated with liquid nitrogen) and reimplanted, resulting in a perfect match for the osseous defect at the surgical site. This process is known as autografting, and it can be very cost-effective. However, there are only limited indications for autografting, as donor bone for allograft is relatively easy to procure.[9][11]
- Metallic Prosthetics
- Metallic prosthetics have revolutionized surgical reconstruction. So-called “mega prostheses” provide for the replacement of large segments of the bone and the joint that connects them. A decade ago, most of these devices had to be custom made but today, “off the shelf” options are available for immediate implantation. Some of these prostheses are expandable “growing” implants that permit interval lengthening. These are particularly efficacious in skeletally immature individuals. Because the growth plates of the affected segment of bone often get resected, the prosthesis can be elongated by 1 to 2 cm at a time, so the length of the previously diseased limb correlates with the contralateral healthy limb.[9][11]
- Tissue Regeneration
- Tissue regeneration for reconstruction following resection of osteosarcoma is a relatively new field. In general, this process utilizes a combination of a patient’s own cells, purified intrinsic growth factors, and synthetic, scaffold-like matrix materials to induce autologous tissue regeneration. Until this emerging technology is more widely available, procedures such as the Ilizarov technique or spatial frame method utilize external fixation devices to promote the growth of long bones up to 1 millimeter per day (about 1 inch per month).[9][11]
Amputation
- Amputation, previously considered the gold standard for surgical management of osteosarcoma, is reserved only for non-resectable masses with contamination of myotendinous and neurovascular that make limb salvage impossible. Amputation may be performed as a standalone treatment or in conjunction with rotationplasty.[9][11]
- Rotationplasty is a procedure that involves resection of the lower extremity to the level of the distal femur. Resection is followed by a 180-degree rotation of the lower extremity with subsequent reattachment at the distal femur, essentially transforming the ankle into a “knee” joint such that the plantar flexors (soleus and gastrocnemius) get converted to knee extensors. The procedure has been shown to provide surprisingly favorable functionality. However, its unusual appearance has been known to cause psychological distress in some patients.[9][11]
Outcomes of Limb Salvage vs. Amputation
- A few studies have demonstrated a slight increase in the rate of recurrence in patients with limb salvage when compared to amputees. Still, the overall rate of survival in patients who recur is comparable. Interestingly, though, several studies report higher survival rates in patients who have undergone limb salvage versus amputation. The vast majority of providers who treat osteosarcoma now favor limb salvage over amputation.[9][11]
Radiation Oncology
National Comprehensive Cancer Network's 2020 Guidelines of Radiation Therapy for Osteosarcoma (Version 1.2020)
Post-Operative Radiation Treatment for Primary Tumors
- Resectable Tumors- Post-operative radiation therapy with 55 Gy plus 9 to 13 Gy boost for residual microscopic or gross disease (64-68 total dose)
- Unresectable Tumors- 60 to 70 Gy (the total dose is dependent on tolerance on normal tissue)
Radiation Treatment for Metastatic Disease
- Samarium 153-EDTMP
- Stereotactic radiosurgery (SRS)
Medical Oncology
Before the advent of chemotherapy, the survival rates for patients with high-grade osteosarcoma were abysmal despite the removal of all visible disease via amputation; this indicated the presence of undetectable micrometastases, typically to the lungs. Chemotherapy, in conjunction with surgical resection, has addressed the presence of micrometastases and significantly improved survival rates.[12]
National Comprehensive Cancer Network's 2020 Recommendations for Chemotherapy Agents & Regimens for Treatment of Osteosarcoma (Version 1.2020)
- Recommended Neoadjuvant/Adjuvant Chemotherapy Regimens for Initial-Occurrence
- Cisplatin and doxorubicin (Category 1)
- MAP (high-dose methotrexate, cisplatin, and doxorubicin) (Category 1)
- Doxorubicin, cisplatin, ifosfamide, and high-dose methotrexate
- Recommended Chemotherapy Regimens for Relapsed, Refractory or Metastatic Disease
- Regorafenib (Category 1)
- Ifosfamide (high dose) +/- etoposide
- Sorafenib
- Sorafenib and everolimus
“Category 1” recommendations are those based upon high-level evidence, with uniform NCCN consensus that the intervention is appropriate. This designation represents the highest level of clinical confidence in efficacy.
Staging
Two popular systems exist for the staging of bone tumors. The musculoskeletal tumor society's Enneking system is used primarily by orthopedic surgeons because it takes into account the anatomic location of the tumor: intracompartmental (completely contained within the bone) vs. extracompartmental (extension outside of the bone). The alternative system described by the American joint committee on cancer (AJCC) does not take anatomic location into account. However, it does account for the size of the tumor, which research has recognized as having significant prognostic value for predicting response to treatment and overall survival. Specifically, larger lesions have a propensity to metastasize, so these patients may benefit from chemotherapeutic intervention, making the AJCC system more popular with oncologists.[13]
Musculoskeletal Tumor Society/Enneking System for Staging of Malignant Musculoskeletal Tumors[8]
- Stage IA: Low grade, intracompartmental tumor location, no metastasis
- Stage IB: Low grade, extracompartmental tumor location, no metastasis
- Stage IIA: High grade, intracompartmental tumor location, no metastasis
- Stage IIB: High grade, extracompartmental tumor location, no metastasis
- Stage III: Any grade, any location, metastasis present
American Joint Committee on Cancer (AJCC) System for Staging of Primary Bone Sarcomas (8th Edition)[14]
- Stage IA: Low grade, less than 8 cm tumor size, no spread to regional lymph nodes, no distant metastasis
- Stage IB: Low grade, greater than 8 cm tumor size or skip lesions, no spread to regional lymph nodes, no distant metastasis
- Stage IIA: High grade, greater than 8 cm tumor size, no spread to regional lymph nodes, no distant metastasis
- Stage IIB: High grade, less than 8 cm tumor size, no spread to regional lymph nodes, no distant metastasis
- Stage III: High grade, discontinuous tumor involvement/"skip" lesions, no regional lymph nodes, no distant metastasis
- Stage IVA: Any grade, any size, no regional lymph node spread, lung metastasis
- Stage IVB: Any grade, any size, regional lymph node spread, lung or extrapulmonary metastasis
Prognosis
The prognosis of osteosarcoma depends on many factors. The following factors dictate the prognosis.
Middle-age patients (over 40 years old) have considerably worse survival rates than younger adults even after the exclusion of secondary forms of osteosarcoma. Several studies have determined that patients over the age of 40 were more apt to have involvement of the axial skeleton and metastatic lesions on presentation, which correlate with poorer outcomes (as described below). Older patients (older than 60 years) fare the worst, typically due to refusal of chemotherapy and radical surgery.[12]
Men reportedly demonstrate less response to chemotherapy, a higher propensity for recurrence, and a four-fold increase in morbidity. Conversely, the female sex correlated with a higher percentage of chemo-related tumor necrosis as well as greater overall survival.[12]
Serum alkaline phosphatase, a biomarker associated with bone turnover, has been found in elevated levels in patients with osteosarcoma. However, it is crucial to consider the age of the patient when interpreting ALP levels as intrinsically higher values are typical in younger age groups. Research has documented high levels in association with less disease-free survival. However, serum alkaline phosphatase levels may be normal at the time of diagnosis in nearly half of patients, particularly in cases where a tumor features minimal osteoid deposition. Lactate dehydrogenase (LDH) is also a useful biomarker. Significantly higher serum LDH levels have been observed in patients with metastasis on initial presentation than patients with local disease alone.[15]
Patients with tumors located in the axial skeleton tend to fare worse compared to those diagnosed in the appendicular skeleton. A difference of up to 10 years of survival exists between groups. Furthermore, patients with femoral tumors often do much worse than patients with lesions located in the distal tibia.[12]
Larger/bulky tumors, as one may expect, carry worse prognoses than smaller lesions. One study found that the morbidity likelihood is 3.4 times higher in larger masses (over 15 cm). When tumor volume exceeds 200 mL, patients are significantly less likely to have successful limb salvage; they also demonstrate a poorer response to chemotherapy and a greater likelihood of recurrence. Unsurprisingly, the chance of death is significantly higher in patients with evidence of metastasis on presentation.[12]
The role of histology in response to chemotherapy and survival outcome is modest. Fibroblastic differentiation is generally considered to be favorable histology. This histologic profile is associated with improved chemotherapy-related tumor necrosis as well as a lower risk of death than alternative histologic subtypes. Chondroid predominant tumor histology correlates with poorer outcomes.[12]
- Preoperative Chemotherapeutic Response
Survival outcome is dependent upon several factors, but the most important predictor of success is the degree of chemotherapy-induced tumor necrosis; necrosis of 90% or more of the tumor is associated with an excellent prognosis.[12]
Osteosarcoma patients have an increased risk of local recurrence and a decreased rate of survival if a pathological fracture is a feature of the initial presentation. Pathological fractures sustained during preoperative chemotherapy have been found to have a decreased rate of survival compared with patients without therapy-associated pathologic fracture.[16]
High BMI has correlations with reduced overall survival.[17]
Complications
Many complications can arise due to osteosarcoma itself or from modalities used to diagnose or treat it.
- Tumor-Specific Complications
Complications of the tumor itself include pathological fractures. These may occur at presentation or during preoperative chemotherapy. As mentioned above, patients in both these scenarios have poorer outcomes than those without pathological fractures.[16]
- Biopsy-Related Complications
When devising an approach to biopsy a lesion concerning osteosarcoma (or any sarcoma, for that matter), careful planning of the biopsy approach is necessary to lower the potential for tumor cells to seed the biopsy tract and surrounding tissues. A biopsy tract that extends across multiple compartments may necessitate a larger field of resection, which increases the risk of treatment-related complications.[18]
- Treatment-Related Complications
- Chemotherapy Side Effects
- When utilizing chemotherapy, the majority of the side effects occur during the treatment process. Nausea, malaise, alopecia, anemia, and anorexia are possible but usually resolve shortly after the completion of the chemotherapy cycle. There are, however, some long-term side effects, which may include cardiotoxicity, pulmonary toxicity, and gradual hearing loss. There are reports of later development of a secondary malignancy.[19]
- Radiation Side Effects
- Radiation is known to impart superficial side effects, including skin dryness, itching, peeling, and uncommonly, burns. Menstrual changes, erectile dysfunction, and infertility are all reported adverse events in cases of pelvic radiation. When the chest and abdomen are involved in radiation treatment, diarrhea, incontinence, rectal bleeding, nausea, vomiting, dry mouth, dysphagia, pneumonitis, and fibrosis are possible. Much like chemotherapy, there is a small risk of late development of a secondary malignancy.[20]
- Periprosthetic Infection
- Prostheses-related infections are a relatively frequent complication (approximately 10% of limb salvage surgeries) most often due to lengthy surgery time, repeated surgery at the same site, and immunosuppression secondary to chemotherapy. First-line treatment of these periprosthetic infections typically involves one or more debridement procedures with both local and systemic antibiotic therapy (systemic and local antibiotic cement beads). If these efforts are ineffective, the implant requires removal, followed by debridement and washout. A cement spacer impregnated with an antibiotic generally gets placed before the insertion of a new prosthesis. Ultimately, amputation may be necessary for a number of these patients.[11]
- Implant Failure
- The most common reason for reconstruction failure is the mechanical breakdown of the mega prosthesis. Mechanical failure necessitates the replacement of the prosthetic. The tibia is the most frequent site of mechanical failure.[11]
- Fracture/Non-Union of Allograft/Autograft
- Fracture/non-union of allograft/autograft reconstruction is a relatively infrequent complication, but it does occur. Chemotherapy, radiation, and extracorporeal treatment of autograft bone have been reported to increase the risk of these complications. Refractory cases may necessitate metallic implant placement or amputation.[11]
Consultations
Management of osteosarcoma requires a multidisciplinary team. Following specialties are involved to improve outcomes.
- Musculoskeletal Radiology (imaging interpretation)
- Orthopedic Oncology (biopsy, resection, limb salvage, prosthetics, grafting)
- Musculoskeletal Pathology (biopsy and surgical pathology histologic interpretation)
- Medical Oncology (preoperative/postoperative chemotherapy and neoadjuvant therapy)
- Radiation Oncology (radiation therapy)
- Physical/Occupational Therapy (post-surgical therapy to regain/improve function)
Deterrence and Patient Education
Symptoms of bone pain, joint pain, and/or palpable mass warrant professional assessment. Patients and their families should be educated on these presenting symptoms as they may potentially be related to an osseous neoplasm. Pain is the major complaint in osteosarcoma, patients must be educated on pain management and available options. Because depression and anxiety are common, counseling of both the patient and the family is necessary. The patient and the family must be educated about the treatment options, pain management, and support services.
Enhancing Healthcare Team Outcomes
Patients with osteosarcoma should ideally be under the management of an interprofessional team, including specialists from radiology, pathology, medical/surgical oncology, and orthopedics. Musculoskeletal radiologists and pathologists are essential for the interpretation of imaging findings and tissue samples for definitive diagnosis and establishing a prognosis. Oncologists are crucial team members who provide the appropriate neoadjuvant and adjuvant chemotherapy treatment and assist with long-term surveillance to monitor for local or distant relapse. A pain specialist is usually involved in helping manage the pain. A board-certified oncology pharmacist should work with the oncologist on agent selection and dosing, as well as educate the patient on pain management and available options. Because depression and anxiety are common, a mental health nurse should be involved in counseling both the patient and the family. The oncology nurse should educate the patient and the family about the treatment options, pain management, and support services as well as assist with coordination of care and follow-up. If the malignancy is advanced, a palliative team should be involved early in the care.
Orthopedic oncologists develop and execute a plan for resection of the tumor, followed by appropriate reconstruction. It is critical to correlate the gross, radiographic, and microscopic findings to establish the correct diagnosis and determine the best course of treatment as osteosarcoma can be highly varied in appearance, imaging features, and histological profile. Following treatment, patients require long-term surveillance because of the possibility of tumor recurrence and extra-osseous metastases. An interprofessional team approach is vital if one wants to improve outcomes and improve the quality of life.[21] [Level V]