Continuing Education Activity
Retinoblastoma is the most common primary intraocular malignancy of childhood. Patients management is best in consultation with an experienced ophthalmologist who will be able to provide appropriate support. To achieve good results, the goals and objectives of retinoblastoma treatment should be defined. This activity demonstrates the use of chemotherapy in the treatment of retinoblastoma, radiotherapy, surgical options, complications, and highlights the role of the interprofessional team in caring for patients undergoing retinoblastoma treatment.
Objectives:
- Describe the indications of retinoblastoma treatment.
- Outline the complications of retinoblastoma treatment.
- Describe the epidemiology and etiology of retinoblastomas.
- Summarize interprofessional team strategies for improving care coordination and communication to advance the treatment of retinoblastoma and improve patient outcomes.
Introduction
Retinoblastoma is an uncommon type of malignancy occurring in 1 per 18000 childbirths,[1] but it is the most commonly encountered primary intraocular malignancy of childhood and accounts for 3% cases of all childhood cancers.[2] It is also the second most prevalent intraocular malignant tumor after uveal melanoma. In specialized care centers, survival rates are up to 95% with retention of vision in most cases, but it is lower in developing countries. Retinoblastoma is composed of retinoblasts (basophilic cells with hyperchromatic nuclei and scanty cytoplasm). Mostly retinoblastomas are undifferentiated, but different degrees of differentiation are present owing to the formation of structures known as rosettes. The tumor can be endophytic (in vitreous) and seeding of tumor cells throughout the eye, or it can be exophytic (in subretinal space), or it can demonstrate a mixed presentation. Optic nerve invasion can occur with the spread of tumor in subarachnoid space and into the brain. Metastatic spread occurs in regional lymph nodes, liver, lungs, bones, and brain.[3]
Etiology
Retinoblastoma occurs as a result of a mutation in the RB1 tumor suppressor gene located at the long arm of chromosome 13 at locus 14 (13q14).[4] Formation of tumor occurs when both the copies of the RB1 gene are mutated. In the case of bilateral retinoblastoma, there are 98% chances that the mutation is germline. Only 5% of cases of retinoblastoma have a family history. 95% of retinoblastoma cases are sporadic, of which 60 % of patients have unilateral disease with no associated germline mutation. Remaining patients present with germline mutations along with the development of multiple tumors.
In this type of retinoblastoma, there is a mutation in one of the alleles of the RB1 gene in all body cells. When the second allele has a mutation as a result of some mutagenic event, it leads to the malignant transformation of cells. Due to the presence of the mutation in all cells, a large number of these children develop bilateral and multifocal retinoblastoma. Heritable disease patients are at significant risk of nonocular cancers such as pineoblastoma, osteosarcoma, soft tissue sarcomas, and melanomas: these malignancies usually occur in a particular age group.[5] The chances of second malignancy are 6%, but the risk increases five-fold when external beam radiation has been used to treat the primary tumor.
Non-heritable retinoblastomas are unilateral and are not transmitted. There is no risk of non-ocular cancers in these patients. In the case of unilateral retinoblastoma with no positive family history, it is non-heritable retinoblastoma, and the corresponding risk in each sibling and patient's offspring is 1%. Almost 90% of unilateral retinoblastomas cases are of the nonhereditary form.
Epidemiology
Retinoblastoma is the most commonly encountered primary intraocular tumor of childhood and accounts for 3% of all childhood tumors. It is also the second most common intraocular malignant tumor. The number of retinoblastoma cases ranges from 1 in 14000 to 1 in 20000 live births.[6] Three hundred new cases occur in the US per year. Retinoblastoma occurs equally in both sexes, and there is no sexual preference. Ninety percent of the cases present before the age of three years. Incidence of the disease is different in the various geographical regions.[7] Research shows six cases per million in Mexico and four cases per million in the US. India and Africa have the highest incidence.[2]
History and Physical
Presentation
Patients with retinoblastoma present mostly within the first year of age in the case of bilateral disease and within 3 years of age in case of unilateral disease. It is important to ask about family history of ocular malignancies. The most common presenting features are the following:
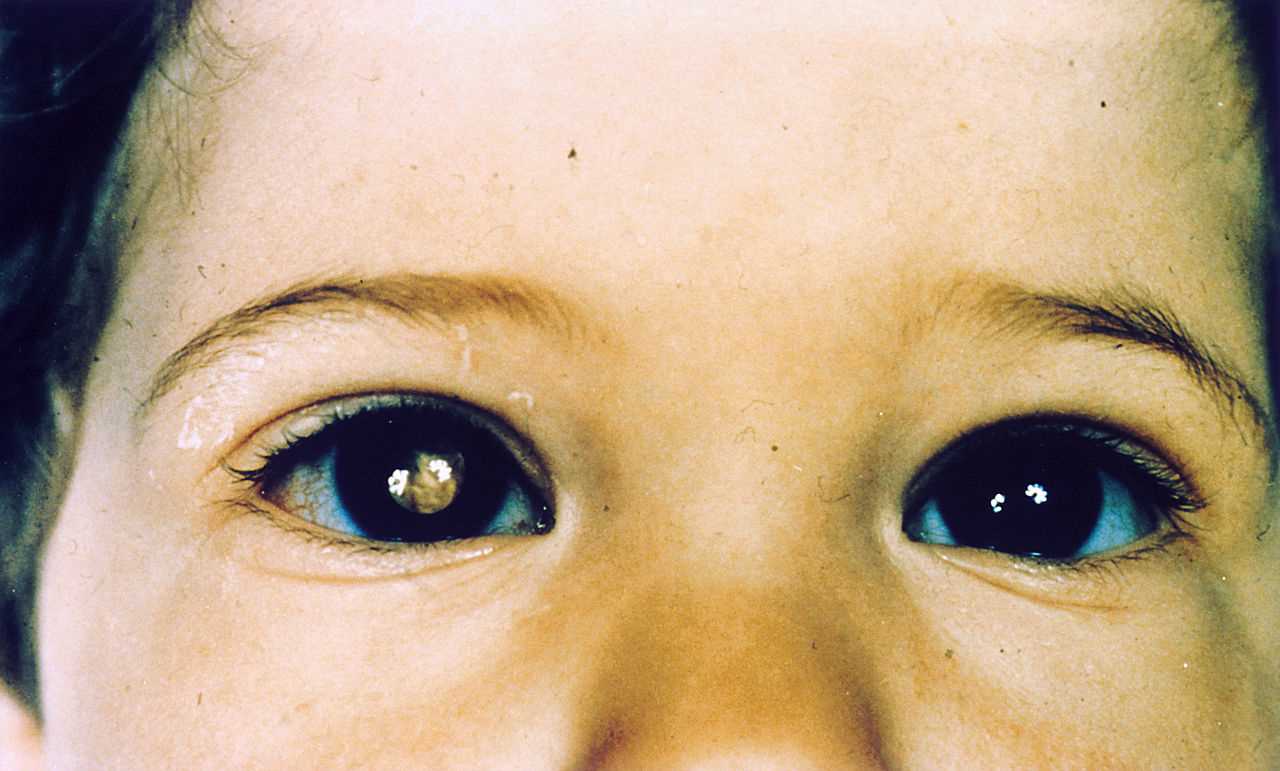
- Leucocoria: (whitish pupillary reflex): It is the most common presenting feature and accounts for 60% of cases.
- Strabismus: It is the second common presenting feature, and it is therefore important to perform fundus examination in all patients of childhood squint.
- Painful red eye: Painful red along with secondary glaucoma and associated buphthalmos can be present.
- Inflammation: Orbital inflammation resembling pre-septal or orbital cellulitis can also be a presenting feature.
- Visible extraocular growth
- Restriction of extraocular movements
- Metastatic disease: Metastatic disease involving lymph nodes, liver, lungs, brain, and bones is rare before ocular involvement.
Signs
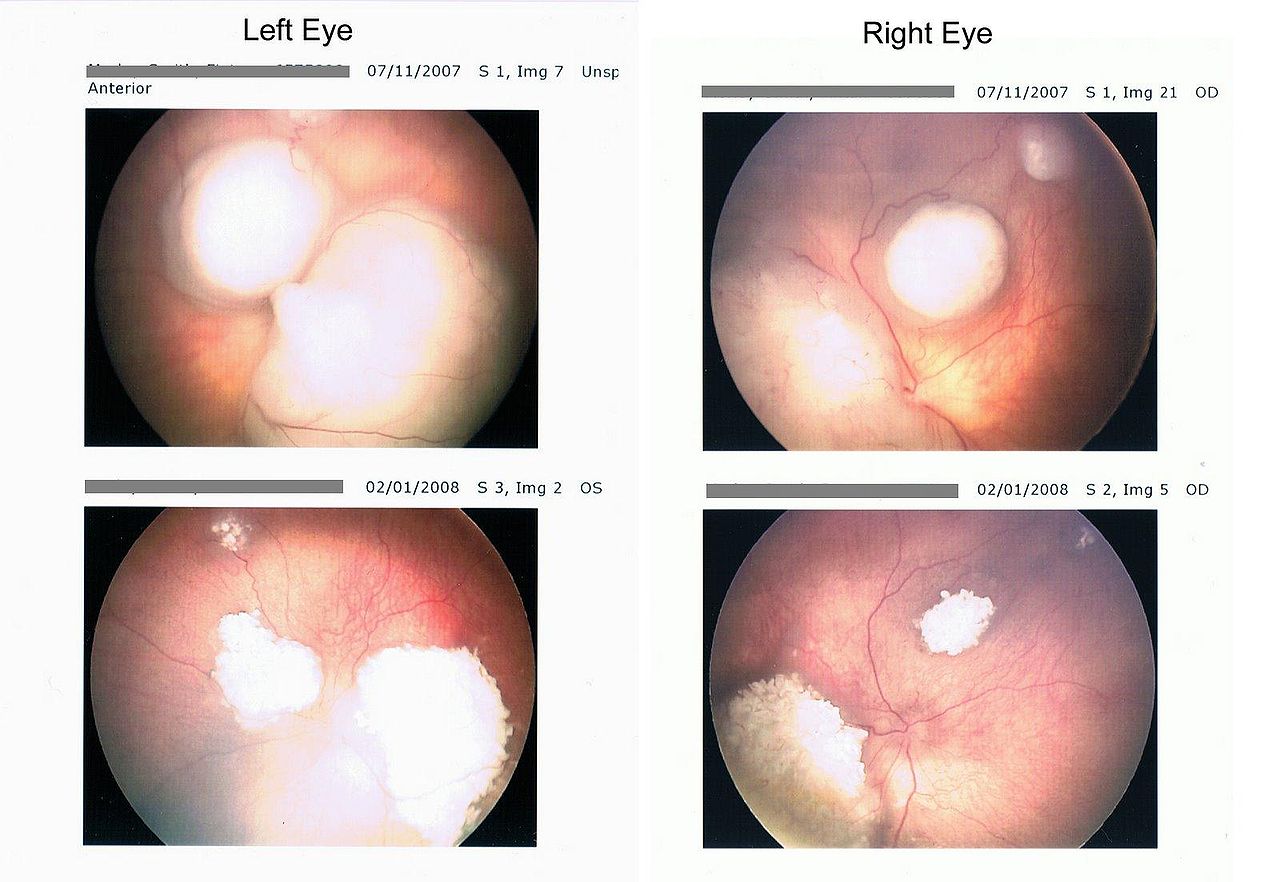
- Intraretinal tumor: Intraretinal retinoblastoma is homogenous, dome-shaped whitish lesion along with calcification.
- Endophytic tumor: The endophytic tumor is present in vitreous as whitish lesion and seeds in the gel.
- Exophytic tumor: Presents as whitish subretinal mass, and it causes retinal detachment.
Evaluation
Red reflex testing with a direct ophthalmoscope is the simplest test, and leukocoria is easily observable. This method serves as a simple screening test.[8]
- Examination Under Anesthesia
Examination under anesthesia is necessary for measuring the corneal diameter, for tonometry, anterior chamber examination with a hand-held slit lamp, fundoscopy, cycloplegic refraction, and documenting all findings.
To assess the size of the tumor, to observe calcifications, and it also helps to rule out similar conditions like coats disease.
Wide-field photography is used for analysis, documentation, and helps in the management of retinoblastoma.
CT scans help in the detection of calcifications, but due to radiation risks, it is avoided upon making the primary diagnosis.
MRI is useful in the evaluation of optic nerve, extraocular extension, pineoblastoma, and to exclude similar diseases.[9]
This includes physical examination, MRI orbit and brain, bone scan, bone marrow aspiration, and lumbar puncture.
Genetic studies of blood samples and tumor tissue from patient and relatives.[3][8]
Treatment / Management
Treatment of retinoblastoma involves a multidisciplinary approach involving an ophthalmologist, pediatric oncologist, ocular pathologist, geneticist, allied health professional, and parents. Different treatment modalities employed in the treatment of retinoblastoma are;
CHEMOTHERAPY is the mainstay of treatment. It is also used in combination with local therapies. Intravenous carboplatin, etoposide, and vincristine are used in three to six cycles depending upon the grade of retinoblastoma. Single carboplatin or dual agent therapy can also be used and has shown favorable results in selective patients such as bridging therapy to avoid aggressive measures. Intravitreal melphalan is used in cases of vitreous seeding although it carries a small risk of extraocular dissemination. Chemoreduction is followed by cryotherapy or transpupillary thermal therapy to maximize tumor control.
TTT (Transpupillary thermal therapy)is used mostly for focal consolidation after chemotherapy; however, it can be used as an isolated treatment. TTT has a direct effect but also augments the effects of chemotherapy.
CRYOTHERAPY: the triple freeze-thaw technique is an option for pre equatorial tumors without deeper invasion or vitreous seedings.
BRACHYTHERAPY is used for an anterior tumor when there is no vitreous seeding and in cases of resistance to chemotherapy.
EXTERNAL BEAM RADIOTHERAPY is avoided when possible, especially in the case of heritable retinoblastoma because it can result in a second malignancy. Retinoblastomas are radiosensitive, but adverse effects include cataract, radiation neuropathy, radiation retinopathy, and hypoplasia of orbit.[10]
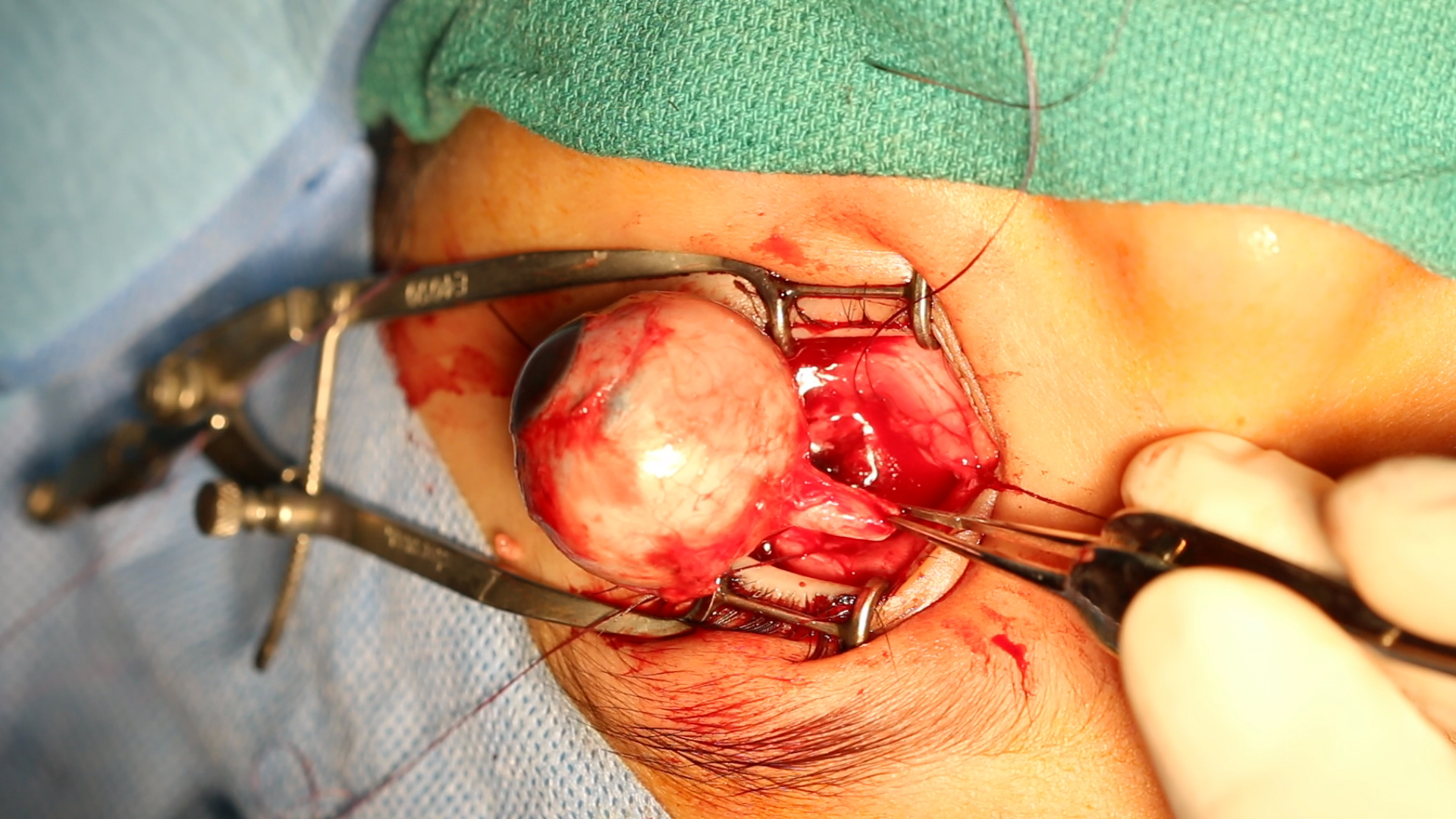
ENUCLEATION: enucleation is performed when there is infiltration of the anterior chamber, neovascular glaucoma, invasion of the optic nerve, and if the tumor comprises more than half of the vitreous volume. It is also useful when chemotherapy has failed and in cases of diffuse retinoblastoma due to poor visual prognosis and a high risk of recurrence. Minimal manipulation should take place when performing enucleation, and a portion of the optic nerve of about 10 mm requires excision.[11] Recent advances in enucleation techniques now allow the removal of a long segment of the optic nerve under direct vision.[12]
EXTRAOCULAR EXTENSION Adjuvant chemotherapy for 6 months is given following enucleation when there is retrolaminar or massive choroidal spread. When the extension of the tumor is up to the cut end of the optic nerve at enucleation, or it is through the sclera, then external beam radiation is used.
REVIEW careful follow-up at repeated intervals is required after treatment for early diagnosis of recurrence or development of new tumor, especially in patients with inherited disease.
Differential Diagnosis
The differential diagnosis of retinoblastoma includes diseases such as:
- Persistent anterior fetal vasculature
- Persistent posterior fetal vasculature
- Coats disease
- Retinopathy of prematurity
- Toxocariasis
- Uveitis
- Vitreoretinal dysplasia
- Coloboma of the choroid and optic disk
- Posterior cataract
Prognosis
Patients with intraocular retinoblastoma, particularly those who have access to modern health care facilities, have an excellent prognosis and an overall survival rate of more than 95% in developed countries. The most critical risk factor associated with poor prognosis is extraocular extension either through the sclera or through the invasion of the optic nerve. Patients who survive bilateral retinoblastoma are at an increased risk of developing non-ocular malignancies later in life the latent period for the development of the second tumor is usually 9 months. External beam radiotherapy decreases the latent period and increases the risk of the second malignancy in the first 30 years of life. The most prevalent type of second malignancy is a sarcoma. The survival of patients who have developed sarcoma is less than 50%.
Complications
If retinoblastoma is left untreated patient is likely to develop the following complications:
- Retinal detachment
- Retinal necrosis
- Orbital invasion
- Optic nerve invasion
- Blindness
- Intracranial extension
- Secondary neoplasms
- Metastasis
- Tumor recurrence
- Temporal bone hypoplasia
- Cataract
- Radiation neuropathy
- Radiation retinopathy
Enhancing Healthcare Team Outcomes
Retinoblastoma management is complex and requires an interprofessional team approach that includes an ophthalmologist, pediatric oncologist, ocular pathologist, geneticist, allied health professionals, and parents. In almost every case, the patient first presents to the nurse practitioner or primary care provider. Since this is a rare disorder that needs urgent attention, the patient should immediately obtain a referral to an ophthalmologist.
The cornerstone of long term retinoblastoma management is strict adherence to the treatment plan, and a careful follow up at repeated intervals is required after treatment for early diagnosis of recurrence or development of new tumor especially in patients with inherited disease.
Patients require education about the type of treatment, including chemotherapy, surgery, and radiation. The primary care providers should be familiar with the postoperative follow up of these patients and when to refer the patient back to the ophthalmologist.
Since chemotherapy is the mainstay of treatment, the involvement of a pharmacist with experience in oncologic treatment is essential. The pharmacist can verify agent selection, dosing, and perform medication reconciliation, reporting any concerns back to the healthcare team. Since nursing is most likely to be in charge of administering the chemo, a close rapport and open communication between pharmacy and nursing staff are crucial.
All patient management is best in consultation with an experienced ophthalmologist who will be able to provide appropriate support. For patients who adhere and to treatment plan and proper follow up, the prognosis is good.
Retinoblastoma is a challenging diagnosis to manage, and as such, requires an interprofessional team approach, including physicians, specialists, specialty-trained nurses, and pharmacists, all collaborating across disciplines to achieve optimal patient results. [Level 5]