Continuing Education Activity
Acute porphyrias are a group of rare disorders characterized by an enzymatic defect in the heme biosynthetic pathway. Patients present with acute, debilitating, and at times life-threatening symptoms that may be precipitated by medications, hormonal changes, starvation, or other factors. This activity covers the clinical presentation, diagnosis, and management of acute porphyria and highlights the role of the interprofessional team in caring for the patient with the condition.
Objectives:
- Describe how a patient with an acute porphyria might present.
- Explain how to evaluate a patient with a suspected acute porphyria.
- Identify a simple, qualitative bedside test that can be used to identify porphobilinogen.
- Explain why an interprofessional team that provides an integrated approach to the care of a patient with acute porphyria can help to achieve the best possible outcomes.
Introduction
Acute porphyria refers to a group of rare disorders characterized by an enzymatic defect in the heme biosynthetic pathway.[1][2] Patients present with acute debilitating, life-threatening attacks that may be precipitated by medications, hormonal changes, starvation, and other factors. In this review, we cover the clinical presentation, diagnosis, and management of acute porphyria.
Etiology
Acute porphyrias are genetic conditions; however, environmental, e.g., drugs and physiological factors, are crucial for clinical manifestation. Porphyria is a genetic disorder. Phenotypic manifestation of the genetic defect is variable and is more common in familial cases. [3] A 50% decline in aminolevulinic acid dehydratase activity occurs in 2% of the general population. Although, the deficiency requires more than 90% inhibition of aminolevulinic acid dehydratase.
More than 100 mutations have been identified. Clinical manifestation is associated with a 50% or more reduction in enzyme function. Porphobilinogen deaminase has three patterns of mutation:
- Type 1: Single-base error causing an amino acid substitution
- Type 2: Confined to the tissue isoform of the enzyme
- Type 3: Deletion in exons
Hereditary coproporphyria is caused by significant heterogeneity and abnormal functioning of coproporphyrinogen oxidase. This is what makes routine genetic screening near to impossible. In heterozygous and homozygous individuals, the reduction in the enzyme is 50% and 90-98%, respectively.
A 50% reduction in protoporphyrinogen oxidase is present in all the samples tested. Mental retardation is prevalent in the neonatal form of variegate porphyria.
Epidemiology
The autosomal dominant acute porphyrias affect males and females equally. Acute porphyria presents in all ethnic and racial groups worldwide.[1][4] Due to the founder effect, certain populations have an unusually high prevalence of porphyrias. Examples of these are the Afrikaner (original settlers from the Netherlands) population in South Africa, who have a high incidence of variegate porphyria (VP), and northern Scandinavian populations with increased numbers of acute intermittent porphyria (AIP). The prevalence of AIP has been estimated to be 5-10 cases per 100,000.[4] The prevalence of acute porphyrias is higher in women, and that reflects the significant exacerbation of their sex hormones. Most patients present after puberty, but the disease manifestation can occur in childhood.
Pathophysiology
Heme is a 64 kDa tetrameric protein present in hemoglobin, myoglobin, respiratory cytochromes, and cytochrome P450 enzymes.[1] The heme biosynthetic pathway involves the conversion of the substrates glycine and succinyl coenzyme A to heme. The pathway consists of 8 enzymatic steps, four enzymes are present in the cytosol, and four enzymes are present in the mitochondria.
The rate-limiting enzyme in the formation of heme is the hepatic ALA synthase1 enzyme, which heme inhibits. The enzyme ALA dehydratase then catalyzes the conversion of ALA to porphobilinogen (PBG). Porphobilinogen deaminase converts PBG to hydroxymethylbilane. Uroporphyrinogen converts this to uroporphyrinogen III, which is then acted upon by uroporphyrinogen decarboxylase to form coproporphyrinogen-III; this undergoes oxidation to protoporphyrinogen IX. The last step is the conversion of this to heme in the presence of iron by ferrochelatase.[5]
Porphyrias fall into two classes: either erythropoietic or hepatic forms depending on the site of the major enzyme deficiency. The enzyme deficiencies are inherited as either autosomal dominant (AD), autosomal recessive (AR), or less commonly as X- linked.[2]
The liver is the source of the four acute porphyrias and also to one of the cutaneous porphyrias (PCT). The most common acute porphyria is acute intermittent porphyria (AIP). AIP is an autosomal dominant acute hepatic porphyria. The enzyme affected is porphobilinogen (PBG) deaminase also referred to as Hydroxymethylbilane synthase (HMBS.)[1][2] Hereditary coproporphyrin (enzyme defect coproporphyrinogen oxidase) and variegate porphyria (enzyme defect protoporphyrinogen oxidase) are also autosomal dominant inherited porphyrias and both present with cutaneous manifestations. AIP, VP, and HCP have a 50% deficiency of the respective enzymes and show low penetrance; thus, around 90% of heterozygotes are asymptomatic for life. The rarest acute porphyria, which is caused by a defect in the enzyme ALA dehydratase, is known as delta-aminolevulinic aciduria or ALA dehydratase deficiency (ALAD) and has an autosomal recessive inheritance pattern.[1][4]][5]
Pathophysiology of symptoms:
The pathogenesis of neurovisceral symptoms in acute porphyria has not been fully elucidated. One hypothesis is that the deficiency of heme affects neuronal function; the other hypothesis is that the precursors aminolevulinic acid (ALA) and porphobilinogen PBG may have direct neurotoxic effects.[1][5]
History and Physical
Patients present initially in adolescence or young adulthood. Attacks in females often have associations with the onset of menstruation. Acute attacks typically consist of severe abdominal pain, nausea, constipation, palpitations, sweating, confusion, and other neurological manifestations such as peripheral neuropathy, seizures, and paresis, tachycardia, and hypertension.[1][2][6]. Psychiatric manifestations, which are present in up to 80% of acute attacks, include behavior change, agitation, depression, hallucinations, altered mental status, and acute psychosis[2]. The most frequent cause of admission in acute porphyria is acute abdominal pain.[2] The patient may also report darkening of the urine (red color), particularly on exposure to light.
Detailed drug history is essential to exclude the intake of precipitating agents. A family history of porphyrias, use of hormonal therapy, and menstrual history in females are also important to elicit on history taking. Online drug databases that provide information about the safety of different drugs in porphyria are available. It is best to refer to the website: www.porphyriafoundation.com/drugs for details. History of recent alcohol intake is also important to note as reports exist of this as a trigger factor for acute attacks.
On examination, the patient may be hypertensive with tachycardia or have evidence of arrhythmias. Motor paresis or poor respiratory effort may be present. Evidence of peripheral neuropathy is common in acute hepatic porphyrias.[2]
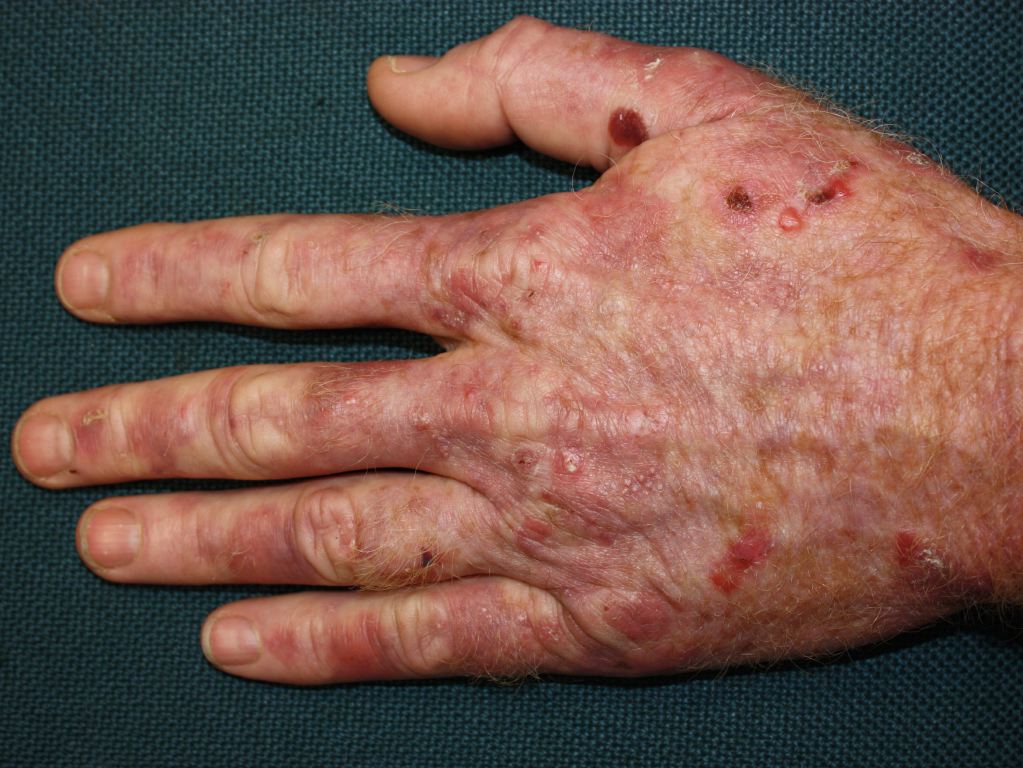
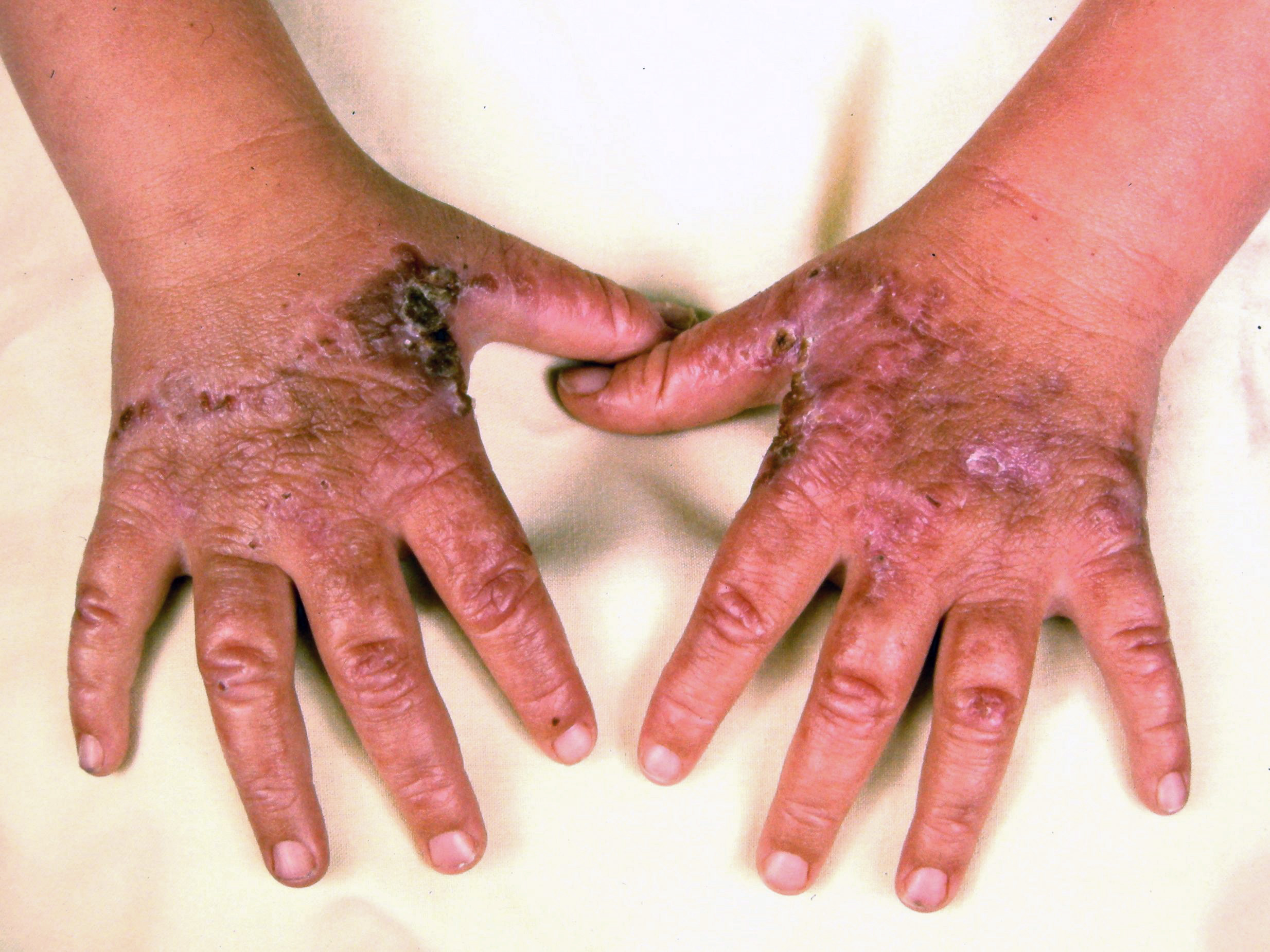
Cutaneous manifestations may be found with certain types of acute porphyrias such as variegate porphyria and HCP.[2] The skin lesions include bullous-type lesions and erosions occurring in sun-exposed areas.[1][7]
Evaluation
Due to the significant degree of overlap with other conditions and varied range of presentation, it is essential to have a high index of suspicion when making the diagnosis. A triad of the following symptoms: visceral abdominal pain, neurological dysfunction, and psychiatric disturbances suggest acute porphyria.[2]
The cornerstone of the diagnosis of acute porphyrias remains biochemical testing. The profile of heme precursors and porphyrins in urine, feces, or blood show a distinct pattern for the different porphyrias. Precursor concentrations are raised in acute attacks but may be normal in periods of remission or asymptomatic individuals.[7] The different porphyrins have different solubilities, and thus some may occur predominantly in urine and others in feces.
In acute attacks, routine biochemical investigation analysis may show the presence of hyponatremia. Additionally, urea and creatinine may be elevated either due to dehydration or renal impairment. Mild elevation of transaminases is common. White cell count is generally normal in the absence of concomitant infection.[5]
The acute porphyrias are characterized by an increase in the intermediate precursor porphobilinogen (PBG). Elevated PBG levels may present around tenfold or greater.
Fresh random urine samples are useful for screening. The use of Ehrlich's reagent in the Watson-Schwartz test provides a bedside qualitative assessment of the presence of PBG. The performance of the quantitative analysis of PBG in urine is by spectrophotometric or chromatographic analysis. In AIP, PBG is usually ten times the upper limit of normal.[5] Occasionally PBG levels may be elevated in genetic carriers who are asymptomatic. Urine PBG may return to normal a few days to weeks after the initial acute attack; hence it is imperative that samples be collected and sent for analysis as soon as acute porphyria is suspected. If there is suspicion of ALAD deficiency, then the measurement of ALA is in order, as this condition will show increased ALA as compared with other acute porphyrias, but it is otherwise unnecessary.
Once the diagnosis of acute porphyria has been made, the next step is to determine which type of acute porphyria is present. High-performance liquid chromatography (HPLC) can identify the different urine porphyrins. Additionally, total fecal porphyrin analysis via spectrophotometry, a fractionation with HPLC/paper chromatography, is used to distinguish AIP from HCP. Fecal porphyrins are generally within normal limits in AIP. In HCP, coproporphyrins are increased in both urine and feces, and this rise is greater than protoporphyrins in feces. In VP, protoporphyrin and coproporphyrin show increases in fecal analysis with a major rise in protoporphyrins. Plasma fluorescence scanning via a fluorescence spectrophotometer is also used to determine the type of porphyria with VP showing a characteristic emission peak at 624-627 nm. The enzymatic analysis may also be performed to determine the enzyme affected.[1][7]
All urine, plasma, and fecal samples must be protected from the light on collection and transportation before analysis.
Genetic testing for mutational analysis can be used to assist the diagnosis in an individual or to screen family members. However, the failure to find a mutation in an individual with a diagnostic history of active acute porphyria does not exclude the diagnosis.[8] Genetic counseling must accompany any such testing.[1]
Treatment / Management
Initial management is primarily directed at managing or eliminating factors such as medications, caloric deprivation, and dehydration that may have precipitated the attack. As such, ensuring rehydration using IV normal saline, glucose infusions, and cessation of any suspected inducer medications is a vital part of the management of the acute attack. Pain relief also forms a part of the initial management, and opioids are considered safe. Definitive treatment involves the administration of IV hemin for 3-14 days, which reverses the increase in ALAS1. Studies are being conducted with small interfering RNA (siRNA) to ALAS1 and appear promising. Givosiran was approved by the FDA for the management of acute hepatic porphyrias in adults.[1][5][4]
Hematin is the only heme-associated compound that is currently approved for use in the United States. Heme arginate is more suitable as it has a low frequency of adverse effects. It has been successfully used in Europe and South Africa.[9]
Ongoing management: some individuals may have recurring attacks. This scenario most often occurs in females and is related to menstruation. These patients should receive a specialist porphyria service referral. Gonadotropin-releasing hormone analogs have been used to prevent ovulation in women that present with recurrent pre-menstruation related acute porphyria. A small number of patients with recurrent attacks have also had liver transplantation.[4][5]
Acute seizure control can be a challenging task in acute porphyrias as most anticonvulsants induce the cytochrome P450 enzyme system. Acute seizure management includes the following:
- First-line drugs: Magnesium sulfate and diazepam
- For status epilepticus: Lorazepam
- Correction of electrolytes: Such as correction of hyponatremia with normal saline considering the volume status of the patient.
- Long-term seizure control: Gabapentin
- Prolonged seizures: Per rectal diazepam
Patients with acute porphyrias tend to have autonomic dysfunction, which can be managed by beta-blockers. An acute rise in blood pressure must be managed with appropriate emergency medication, such as labetalol.
Psychiatric symptoms are controlled by giving phenothiazines, such as chlorpromazine. These medications are also given for nausea in such patients.
Differential Diagnosis
Since the presentation of porphyrias is variable, the list of differential diagnoses is also diverse. Lead poisoning may present with similar symptoms as an acute porphyria attack. There may also be elevated zinc protoporphyrin levels, however, blood lead levels are more specific. Acute abdomen from surgical or gynecological causes as well as acute viral gastroenteritis are other common differentials, but with acute porphyria, there is no rebound tenderness. Other differential diagnoses include:
- Acute anemia
- Hepatitis B and C
- Hodgkin lymphoma
- Acute lymphoblastic leukemia (ALL)
- Pediatric acute myelocytic leukemia
- Pediatric generalized anxiety disorder
Prognosis
Several decades ago, the prognosis for acute porphyria with neurological complications was poor, with a reported mortality of 35%. The prognosis remains guarded for those that present with acute porphyria. However, the number of cases progressing to advanced disease has declined.[5]
Complications
Reports exist of an association of acute porphyria with hepatocellular carcinoma, particularly AIP. AIP also has associations with an increased risk of hypertension and chronic kidney disease.[10] Chronic pain is also a complication of recurrent attacks.
Consultations
A specialist in porphyria should be included in the diagnosis and management of patients with acute and chronic cases of porphyrias. Such specialists may belong to different disciplines, such as gastroenterology, hematology, and metabolic diseases. Consulting a neurologist in case of seizures is advisable. A physical therapist may be needed in cases where muscle weakness develops. Other disciplines to be involved are psychiatry, reproductive medicine, cardiology, and anesthesiologist.
Deterrence and Patient Education
Patients should be made aware of the triggers and alarming signs. A diet rich in carbohydrates can mitigate the illness. The patients should be counseled regarding the essentiality of fluid intake to ensure adequate clearance of porphyrins. In cases where hypertension develops, a low-salt, low-fat, and low cholesterol diet is the best strategy.
Patients should also be advised to restrict activities that may put them at risk for dehydration, carbohydrate depletion, and exhaustion.
Pearls and Other Issues
Porphyrias are largely still underdiagnosed, and the index of suspicion must be high to diagnose. Diagnosis is essential to initiate appropriate treatments as soon as possible.
Enhancing Healthcare Team Outcomes
Because of its diverse presentation, the condition is best diagnosed and managed by an interprofessional team. Biochemical diagnosis remains the crux inherited conditions of acute porphyria. Given the urgency to identify an acute attack, it is essential that laboratory services offer screening tests with a rapid turnaround time. Patient education regarding trigger factors for acute attacks and avoidance of these trigger factors is important. Education regarding the inheritable nature of the disease is important for the patient and family members.