Continuing Education Activity
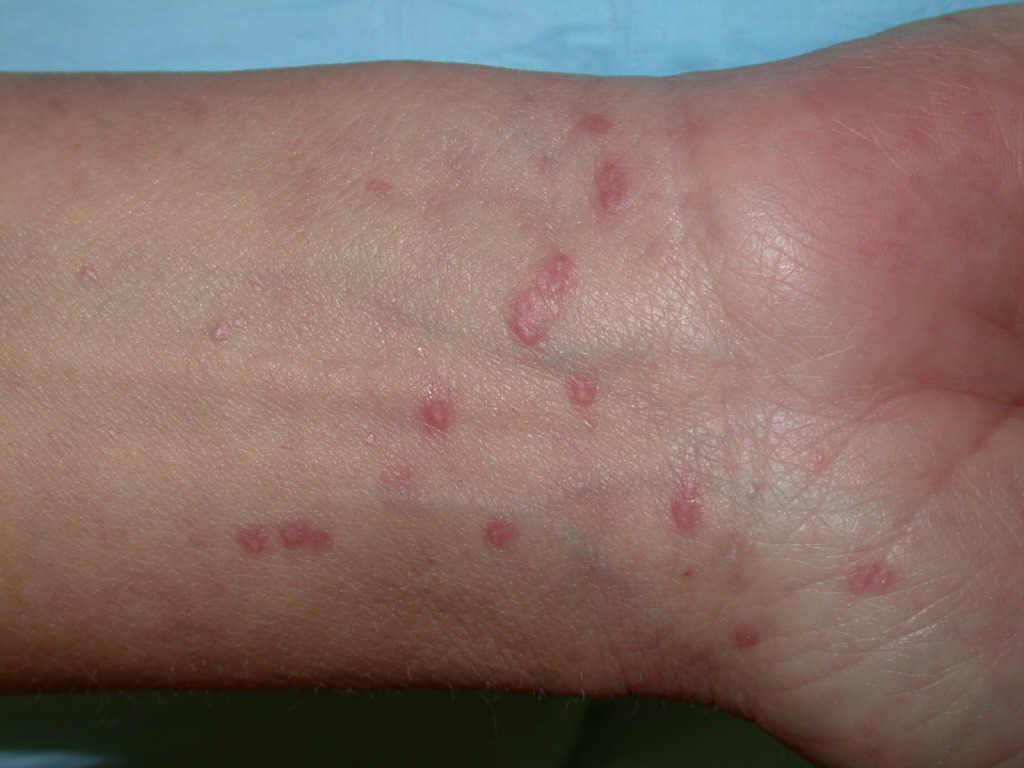
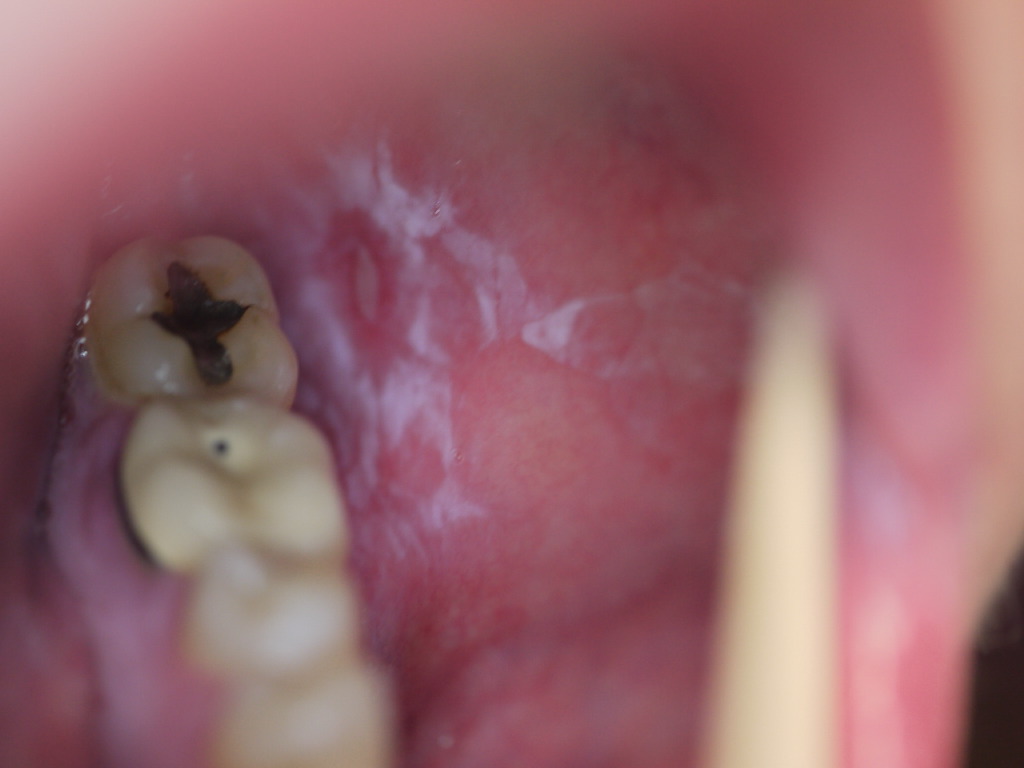
Lichen planus (LP) is an inflammatory disorder of the skin and mucous membranes with no known cause. It appears as pruritic, violaceous papules and plaques most commonly found on the wrists, lower back, and ankles. A lattice-like network of white lines called Wickham striae overlies the lesions but is most easily observed on the buccal mucosa where erosions can also be present. Drug-induced lichen planus, or lichenoid drug eruption, is frequently photo distributed but may be indistinguishable from idiopathic LP. The natural history of LP varies significantly. The majority of patients with cutaneous lesions spontaneously clear within 1 to 2 years after initial presentation. However, recurrences are common, and residual hyperpigmentation of the skin frequently results. By contrast, oral LP is a chronic disease that may or may not remit. Drug-induced LP gradually resolves after removal of the causative medication. This activity describes the interprofessional team's role in the evaluation and management of lichen planus.
Objectives:
Describe the appearance of lichen planus.
Review the causes of lichen planus.
Summarize the types of lichen planus.
Outlne the interprofessional team's role in the evaluation and management of lichen planus.
Introduction
Lichen planus (LP) is an inflammatory disorder of the skin and mucous membranes with no known cause. It appears as pruritic, violaceous papules and plaques most commonly found on the wrists, lower back, and ankles. A lattice-like network of white lines called Wickham striae overlies the lesions but is most easily observed on the buccal mucosa where erosions can also be present. Drug-induced lichen planus, or lichenoid drug eruption, is frequently photo distributed but may be indistinguishable from idiopathic LP.
The natural history of LP varies significantly. The majority of patients with cutaneous lesions spontaneously clear within 1 to 2 years after initial presentation.[1] However, recurrences are common, and residual hyperpigmentation of the skin frequently results. By contrast, oral LP is a chronic disease that may or may not remit.[2] Drug-induced LP gradually resolves after removal of the causative medication.[3]
Etiology
Lichen planus is an idiopathic disease. Its pathogenesis is not fully understood, but it appears to represent a T-cell-mediated autoimmune disease. The prevailing theory is that exposure to an exogenous agent such as a virus, drug, or contact allergen causes alteration of epidermal self-antigens and activation of cytotoxic CD8+ T cells. The altered self-antigens cross-react with normal self-antigens found on basal keratinocytes resulting in T-cell targeting and apoptosis.[4]
A variety of agents have been associated with the development of LP, but a particular note has been made of the link with viruses, especially the hepatitis C virus (HCV). Patients with LP are 5 times as likely to test positive for HCV as the general population, and those with HCV-seropositivity are 2.5 to 4.5 times as likely to develop LP.[5][6]
Oral lichen planus is correlated with contact allergies to a variety of metals found in dental restorations including mercury, copper, and gold. Removal of the sensitizing metal resulting in clearance of LP lesions has been described.[7][8]
A large number of drugs have been associated with LP, but recurrence of lesions following drug rechallenge is rare. More commonly associated drugs include antimalarials, ACEIs, thiazide diuretics, NSAIDs, quinidine, beta-blockers, tumor necrosis factor (TNF)-alpha inhibitors, and gold.[3][9]
Epidemiology
The prevalence of cutaneous LP is approximately 0.2% to 1% of adults worldwide.[10] Oral LP is more common and reported in 1% to 4% of the population. Overall, women are more frequently affected than men at a ratio of 1.5:1, and most cases develop between the ages of 30 and 60. It is rare in children as they represent less than 5% of all LP patients.[5] While LP is not generally considered to have a racial predilection, some recent studies have suggested there may be a higher incidence of the disease in African-Americans and those of Indian and Arabian descent.[11][12] There does appear to be a familial component as up to 10% of first-degree relatives of patients may also develop the disease.[13]
Histopathology
Skin biopsy and microscopic analysis are valuable in confirming the diagnosis in atypical and severe cases as the histopathologic features are largely the same regardless of the distribution or subtype. Principal findings consist of thickening of the stratum corneum without nuclei present (hyperkeratosis without parakeratosis); irregular thickening of the stratum granulosum; destruction of the stratum basale; alteration or loss of rete ridges resulting in a sawtooth appearance; and a dense band of lymphocytes infiltrating the dermis along the dermo-epidermal junction (interface dermatitis). Additionally, idiopathic LP (almost) never has eosinophils whereas drug-induced LP may have eosinophils.
Apoptotic keratinocytes are often seen near the basal layer and are termed colloid or Civatte bodies. Direct staining by immunofluorescence may display colloid bodies with irregular deposits of IgA, IgM, IgG, or C3.[14][15]
History and Physical
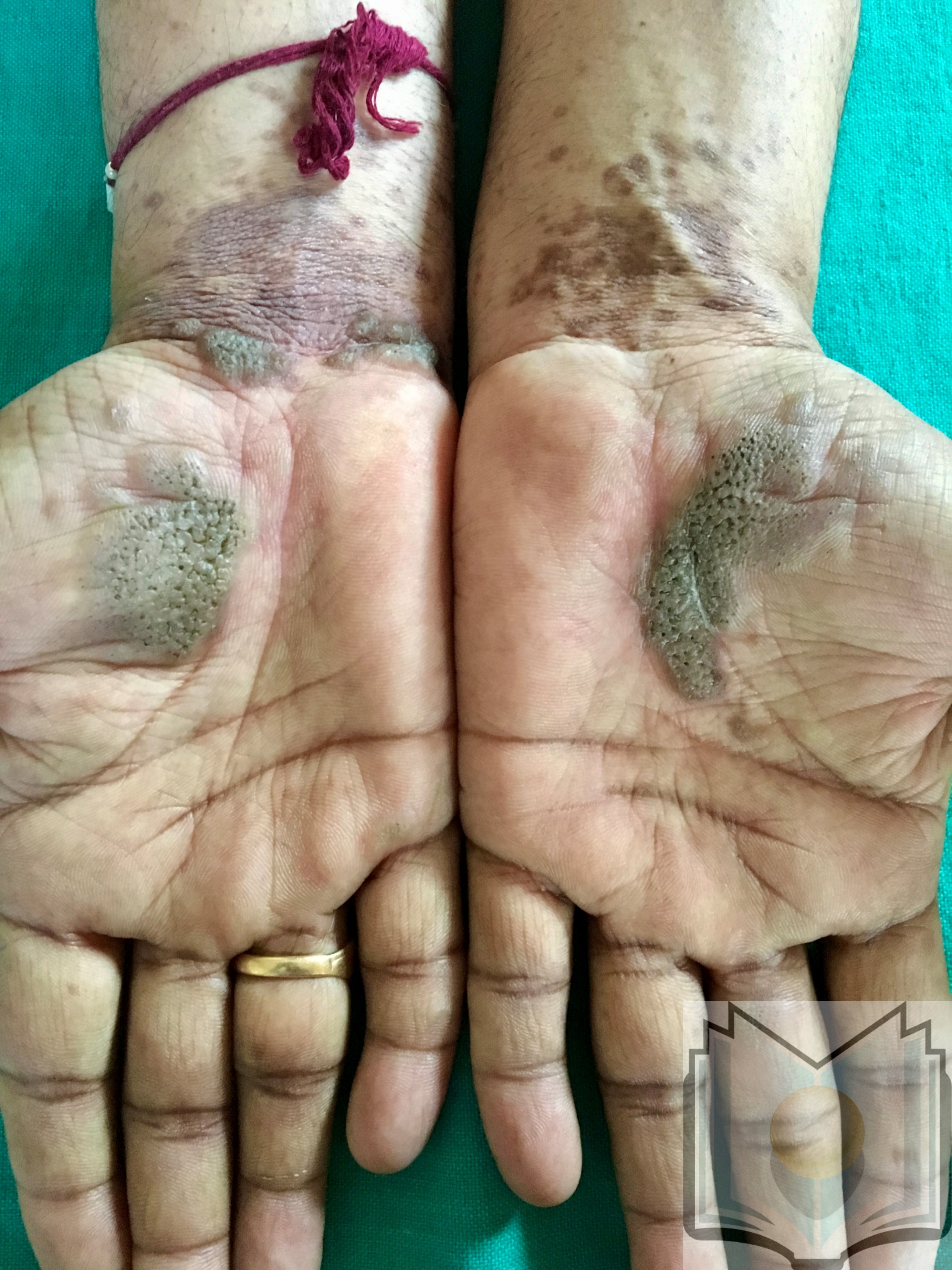
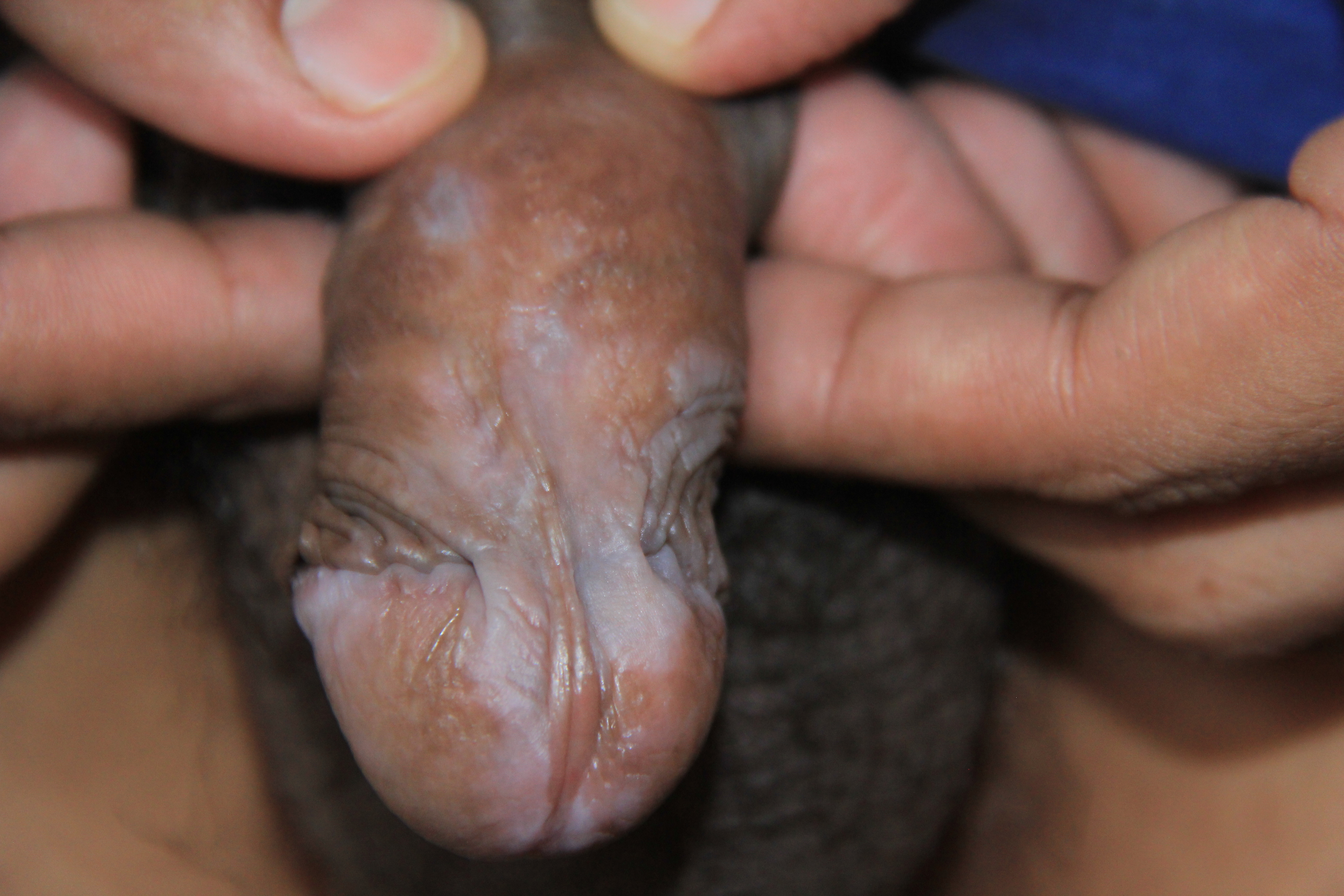
Lichen planus can display a variety of lesion types, but the most common presentation is an area of polygon-shaped, itchy, violaceous, flat-topped papules a few millimeters wide. This classic presentation is known as The Six Ps of LP: purple, polygonal, planar, pruritic papules, and plaques. The lesions have a shiny surface covered in fine white lines known as Wickham striae and are firm on palpation. They may be seen as a few individual lesions, found scattered widely, grouped in plaques, or arranged in annular, linear, or actinic (sun-exposed) patterns. The isomorphic response (i.e., Koebner phenomenon) can be seen in LP wherein new lesions arise in lines where scratching occurs, just as is seen in psoriasis. The most common areas of involvement include the flexor wrists, dorsal hands, lower back, ankles, and shins. Frequently a grayish-brown hyperpigmentation can be found after lesions resolve due to deposition of melanin in the superficial dermis.[5][10]
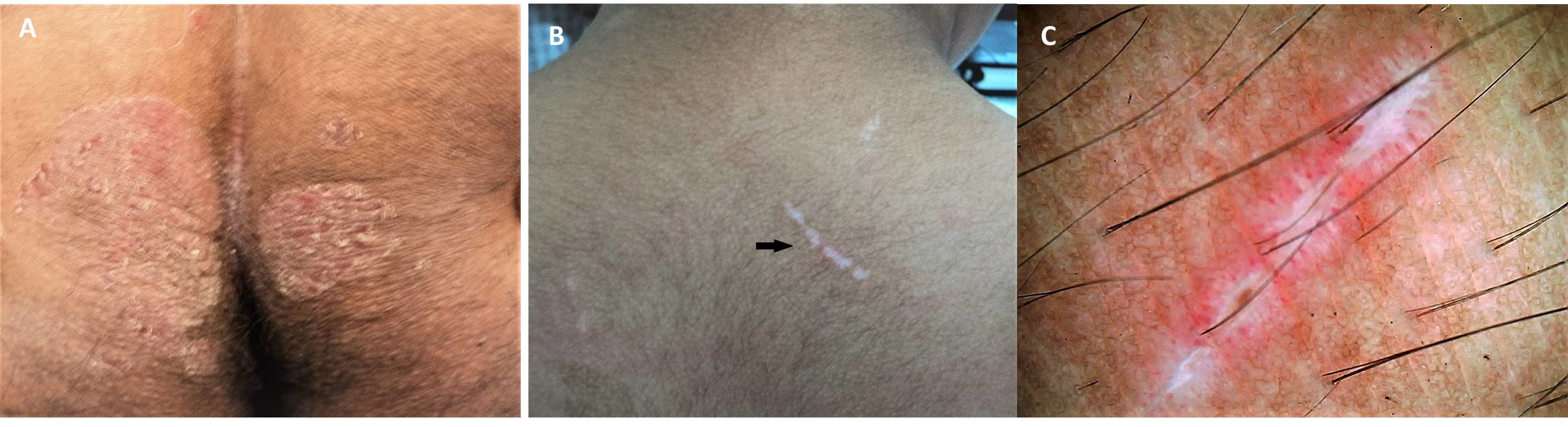
Various subtypes of LP exist that display patterns different from the classic presentation. Hypertrophic LP is often found on the shins and ankles and is characterized by red, red-brown, or yellow-grey papules and plaques that coalesce with a thickened or verrucous surface. Ulcerative LP is found on the soles of the feet or between the toes with painful, erosive lesions that make walking difficult. Bullous LP appears most often on the legs as small to large tense blisters filled with clear or pale-yellow fluid. Lichen planus pemphigoids display features of lichen planus with the development of bullae both on top of LP lesions as well as on unaltered skin. Lichen planus pigmentosus is characterized by development of macular or papular pigmented lesions often arranged in a linear, follicular, or Blashkoid pattern. Inverse LP is analogous to inverse psoriasis in that it is found in intertriginous areas and completely loses its classic appearance. Extensive erythematous lesions with lichenification and without distinct borders are seen in the axillae, limb flexures, inguinal creases, and beneath the breasts.[16]
Mucosal involvement affects over half of all LP patients and frequently is the sole presenting sign. It is most commonly seen in the mouth but can be found on the lips, esophagus, glans penis, vulva, or vagina. Six subtypes of oral LP exist: reticular, erosive, papular, plaque-like, atrophic, and bullous. Reticular is the most common form and presents as asymptomatic white, lacy lines often seen on the bilateral buccal mucosa. The erosive and atrophic forms are commonly associated with a burning pain exacerbated by hot or spicy foods. On the tongue or buccal mucosa, lesions can easily be mistaken for leukoplakia or candidiasis. Esophageal LP predominantly affects women and can give rise to dysphagia, strictures, and possibly squamous cell carcinoma.[17][18][7]
When involving the glans penis, LP commonly displays an annular configuration. By contrast, in women, an erosive variant is most frequently found when LP involves the vulva or vagina, and scarring and strictures are troublesome sequelae that may occur. LP found simultaneously on the female genital and oral mucosa is termed vulvovaginal-gingival syndrome. This severe, desquamative subtype has been associated with the HLA DQB1*0201 gene in 80% of patients suggesting a genetic predilection for its development.[19][20]
LP of the nails occurs in approximately 10% of patients and typically affects multiple nails without necessarily affecting the nearby skin. Thinning of the nail plate and longitudinal ridging are the first signs of disease. Continued involvement leads to scarring of the nail matrix, dorsal pterygium formation, sandpaper nails (trachonychia), and possibly complete loss of the nail plate. Some patients may display a variant of LP called twenty-nail dystrophy wherein such findings on all twenty nails are the only presenting sign of disease. This subtype is much more common in children than adults.[21][22]
Lichen planus of the scalp and other hair-bearing regions is called lichen planopilaris (LPP). Small, red, follicular papules and macules appear where inflammation is present and lead to progressive scarring alopecia. LPP may appear alone or with typical LP lesions elsewhere on the body. When LPP occurs principally on the anterior scalp and eyebrows in older women it is known as frontal fibrosing alopecia. A familial variant of LPP known as Graham-Little-Piccardi-Lasseur syndrome is characterized by scarring alopecia of the scalp, typical cutaneous or mucosal LP, and non-scarring loss of pubic and axillary hairs with follicular papules.[23][24]
Rather than appearing in the classic LP sites, lichenoid drug eruption frequently appears in sun-exposed areas, is symmetric, and is more generalized in distribution. Lesions look more eczematous or psoriasiform, and Whickam striae are not commonly seen. There is typically a latent period of several months to a year from initiation of a drug to an eruption of lesions, so thorough review of medication history is vital to diagnosing drug-induced LP.[3]
Evaluation
While in the clinic, dermoscopy allows visualization of Wickham striae in most cases. A network of white lines with red globules along the periphery is the classic finding.[25]
Oral LP lesions located near dental restorations should prompt patch testing to determine if an allergy to one of the associated metals exists.[26]
Biopsy with the microscopic analysis is the most useful tool to confirm the presence of LP. Lesions have many characteristics as noted previously that typically allow a definitive diagnosis. Adding direct immunofluorescence to histology can be helpful when trying to differentiate between LP and lupus erythematosus (LE).[27]
Treatment / Management
Cutaneous LP typically clears spontaneously within 1 to 2 years, so treatment is aimed at reducing pruritus and time to resolution. For limited LP, first-line treatment is superpotent topical steroids (clobetasol 0.05%) twice daily for 2 to 4 weeks. Inadequate response to topical steroids may be augmented with intralesional steroid injections (triamcinolone 5 to 10 mg/mL). For diffuse LP, first-line treatment is daily oral corticosteroids (prednisone 30 to 60 mg) tapered over 2 to 6 weeks. If no change is seen, second-line therapy should be considered. Second-line therapy may include metronidazole (500 mg twice daily for 3 to 8 weeks), sulfasalazine (500 mg twice daily increased in 500 mg increments every 3 days until 2.5 grams daily is reached, for 3 to 6 weeks), isotretinoin (10 mg twice daily for 2 months), acitretin (30 mg daily for 8 weeks), PUVA, UVB, topical calcineurin inhibitors, or methotrexate (15 mg per week for adults, 0.25 mg/kg per week for children). Third line treatment may include trimethoprim-sulfamethoxazole, griseofulvin, terbinafine, antimalarials, tetracyclines, ciclosporin, mycophenolate mofetil, azathioprine, etanercept, adalimumab, or low-molecular-weight heparin.[28][5]
Oral LP may spontaneously resolve within 5 years, but many cases are chronic and never resolve. Treatment-induced remission is typically followed by relapse. Thus, asymptomatic oral LP should not be treated as the side-effect burden of treatment is high. The goal for treatment of symptomatic oral LP is to heal erosive lesions to reduce pain and allow normal food intake. Patients should be instructed to avoid spicy or acidic foods as well as alcohol and tobacco as these exacerbate symptoms. First-line treatment is very high potency topical steroids three times daily until remission. No improvement after 6 weeks should prompt escalation of therapy. Second-line treatment is oral corticosteroids or application of topical calcineurin inhibitors. Third-line treatment may include cyclosporine, azathioprine, mycophenolate mofetil, or methotrexate.[29][7]
Consideration of drug-induced LP must always be explored prior to starting therapy. Withdrawal of the suspected drug leading to the gradual disappearance of lesions confirms the diagnosis, although it may take some time for lesions to fully resolve. [30]
Differential Diagnosis
Because LP may be a reaction to different exogenous agents such as viruses, drugs, or contact allergens, care should be taken to identify and treat any underlying causes before diagnosing idiopathic LP.
Various diseases that may appear similar to LP include LE, erythema dyschromicum perstans, psoriasis, secondary syphilis, pityriasis rosea, lichen nitidus, graft versus host disease, and keratosis lichenoides chronica. Hypertrophic LP can look very similar to lichen simplex chronicus. Vulvar LP can be difficult to distinguish from lichen sclerosis.
Differentiating between LP and LE can be a challenge when lesions are only present on the scalp or in the mouth, so biopsy with DIF is especially useful in those cases. The simultaneous presence of both diseases has been described in many reports, possibly related to the use of antimalarials in the treatment of LE.[27][31]
Prognosis
Cutaneous LP often clears spontaneously within 1 to 2 years, but residual hyperpigmentation is very common. Oral LP may clear spontaneously within 5 years, but typically it is a chronic disease with a remitting and relapsing course. Hair loss from LPP is permanent. Drug-induced LP lesions clear slowly after the causative medication is withdrawn.
Enhancing Healthcare Team Outcomes
When healthcare workers including the nurse practitioner encounter lichen planus, the patient should be referred to the dermatologist for definitive management. Lichen planus may affect the skin, scalp and the mucosal organs and is a relatively difficult disorder to cure. Cutaneous LP typically clears spontaneously within 1 to 2 years, so treatment is aimed at reducing pruritus and time to resolution. Many drugs are used to treat the skin manifestations with not much difference among them in effectiveness.
Oral LP may spontaneously resolve within 5 years, but many cases are chronic and never resolve.
The pharmacist should always be aware that some cases of LP may be drug-induced and consult with the treating clinician regarding the patient's case. Withdrawal of the suspected drug leading to the gradual disappearance of lesions confirms the diagnosis, although it may take some time for lesions to fully resolve. [30]