Continuing Education Activity
X-linked ichthyosis, also known as steroid sulfatase (STS) deficiency and X-linked recessive ichthyosis, is a recessive, nonsyndromic genetic skin disorder. This condition is caused by a mutation or deletion in the STS gene, which is responsible for encoding the steroid sulfatase enzyme, causing the complete loss of steroid sulfatase enzyme activity. Although X-linked ichthyosis equally affects all ethnic groups and races worldwide, the condition predominantly affects males rather than females, as suggested by its name. Affected patients can normally produce skin cells but cannot shed them correctly, leading to dry skin that accumulates in the form of polygonal scales. The extracutaneous features include asymptomatic punctate corneal opacities, cryptorchidism, and cognitive or behavioral disorders, such as attention-deficit hyperactivity disorder.
The deficiency of steroid sulfatase results in the accumulation of cholesterol sulfate and depletion of cholesterol levels, leading to an abnormal skin barrier and retention of corneocytes. Although definitive treatment does not exist for X-linked ichthyosis, various options are available for long-term management. The primary treatment objectives involve reducing dryness, minimizing scale formation, and enhancing skin appearance without causing irritation. This activity comprehensively reviews the pathophysiology, diagnosis, and management of X-linked ichthyosis, equipping healthcare professionals with the essential knowledge to deliver optimal, patient-centered care to individuals affected by X-linked ichthyosis.
Objectives:
Identify the characteristic features of X-linked ichthyosis, including thick, dry, scaling skin, and recognize extracutaneous manifestations such as asymptomatic punctate corneal opacities, cryptorchidism, and cognitive or behavioral disorders.
Apply knowledge of the genetic underpinnings of X-linked ichthyosis, particularly the STS gene mutation or deletion, to inform accurate diagnoses and genetic counseling for affected individuals and their families.
Select appropriate interventions for long-term management and evidence-based treatment strategies for X-linked ichthyosis, considering individual patient preferences and characteristics to optimize treatment adherence.
Coordinate care efforts among interprofessional healthcare providers from dermatology, ophthalmology, psychology, and genetics to facilitate seamless communication, patient education, and ongoing monitoring to enhance overall outcomes for individuals affected by X-linked ichthyosis.
Introduction
X-linked ichthyosis (MIM #308100), also known as steroid sulfatase (STS) deficiency and X-linked recessive ichthyosis, is a recessive, nonsyndromic genetic skin disorder. This condition is caused by a mutation or deletion in the STS gene, which is responsible for encoding the steroid sulfatase enzyme, causing the complete loss of steroid sulfatase enzyme activity. Although X-linked ichthyosis equally affects all ethnic groups and races worldwide, the condition predominantly affects males rather than females, as suggested by its name. With an incidence of 1 in 2500 to 1 in 6000 males, X-linked ichthyosis is the second most common type of ichthyosis, after ichthyosis vulgaris.
X-linked ichthyosis was first recognized by Dr Wells and Dr Kerr in 1965. Approximately 15% to 20% of individuals with this condition manifest symptoms at birth, which continue to progress, whereas the rest usually develop symptoms over the following weeks. Affected patients can normally produce skin cells but cannot shed them correctly, leading to dry skin that accumulates in the form of polygonal scales.[1][2] Initially, the symptoms are usually mild and advance to the development of large, polygonal, brownish scales. The extracutaneous features include asymptomatic punctate corneal opacities, cryptorchidism, and cognitive or behavioral disorders, such as attention-deficit hyperactivity disorder (ADHD).
The deficiency of steroid sulfatase results in the accumulation of cholesterol sulfate and depletion of cholesterol levels, leading to an abnormal skin barrier and retention of corneocytes. Although scaling typically does not affect flexural surfaces, such as the popliteal and antecubital fossae, palms, soles, hair, and nails, the anterior surface of the lower extremities is typically the most affected. Unfortunately, a definitive treatment does not exist for X-linked ichthyosis; however, various options are available for long-term management. The primary treatment objectives involve reducing dryness, minimizing scale formation, and enhancing skin appearance without causing irritation.
Etiology
The cutaneous scaling observed in X-linked ichthyosis is primarily caused by a complete deletion of the STS gene on chromosome Xp22.31. Partial deletions or point mutations may also contribute. Female carriers of STS gene mutations do not exhibit any disease manifestations, as the gene localizes to a region of the X-chromosome that does not undergo X-inactivation. De novo STS mutation may also occur.[3][4] Approximately 8% of affected patients may have mutations in adjacent genes, leading to additional genetic syndromes depending on the resulting variation. Another subset of patients may carry a filaggrin gene (FLG) variant, contributing to a more severe phenotype and a higher likelihood of developing atopic dermatitis.
Epidemiology
X-linked ichthyosis is the second most prevalent form of ichthyosis, with ichthyosis vulgaris being the most common type. Although this condition equally affects all ethnic groups and races worldwide, the condition predominantly affects males rather than females, as suggested by its name. The incidence of X-linked ichthyosis ranges from 1 in 2500 to 1 in 6000 males. Notably, there have been reports of 3 cases of X-linked ichthyosis in females from carrier mothers and affected fathers.[3][5]
Pathophysiology
Steroid sulfatase is responsible for the hydrolysis of certain alkyl steroid sulfates, such as dehydroepiandrosterone sulfate (DHEAS) and aryl steroid sulfates, converting them to their unconjugated or unsulfated forms. Steroid sulfatase activity within the epidermis is notably high in the granular layer and extends into the stratum corneum. This enzymatic activity is responsible for cholesterol generation, which is essential in forming extracellular lamellar bilayers. These bilayers regulate the permeability barrier function and desquamation process. In cases of steroid sulfatase deficiency, an accumulation of cholesterol sulfate (COSO4) in the stratum corneum inhibits proteases responsible for corneodesmosome degradation. This impairment in desquamation leads to increased corneocyte cohesion, retention hyperkeratosis, and compromised skin permeability.
Steroid sulfatase is a crucial enzyme for estriol synthesis in the human placenta. Steroid sulfatase hydrolyzes conjugated or sulfated alkyl steroid sulfates, such as dehydroepiandrosterone (DHEA), transforming them into their unsulfated or unconjugated form, as exemplified by the conversion of DHEAS→DHEA. Consequently, decreased steroid sulfatase activity leads to elevated conjugated and reduced unconjugated steroid sulfates. Notably, in women carrying an affected fetus, the absence of placental steroid sulfatase activity lowers the mother’s unconjugated serum estriol (uE3). This, in turn, could lead to the diagnosis of X-linked ichthyosis during the prenatal period, identified through the detection of low serum estriol levels in prenatal screening for Down syndrome. The comprehensive effects of steroid sulfatase deficiency on pregnancy and delivery remain uncertain. Some researchers propose that steroid sulfatase deficiency leads to prolonged labor and resistance to pitocin.
Although the mechanism behind the increased incidence of atrial fibrillation is unclear, researchers suspect the involvement of circulating levels of DHEAS. Additional findings associated with steroid sulfatase deficiency include asymptomatic corneal opacities, cryptorchidism, neurological and cognitive development abnormalities, and anosmia when the deletion encompasses more than a gene—a phenomenon known as contiguous gene syndrome.[5] Further details about the specific genetic variations are available online at the Online Mendelian Inheritance In Man (OMIM) website: https://www.omim.org/entry/308100.
Histopathology
In cases with mild symptoms, biopsy results may appear normal. Biopsies from areas with more noticeable scaling reveal compact laminated eosinophilic orthokeratotic hyperkeratosis or thickening of the cornified layer without retained nuclei, accompanied by dermal perivascular and periappendageal inflammation. The thickness of the granular layer may vary, exhibiting thinness, normality, or thickening, and clinicians may also observe mild acanthosis.[6]
History and Physical
Classic X-Linked Ichthyosis
The timing of symptom onset is variable. Approximately 15% to 20% of individuals experience symptoms at birth, whereas others develop symptoms in the subsequent weeks. The scaling usually starts as mild and advances to larger, polygonal, translucent scales during childhood into adolescence. Notably, it is rare for patients affected by X-linked ichthyosis to present with a collodion membrane at birth.
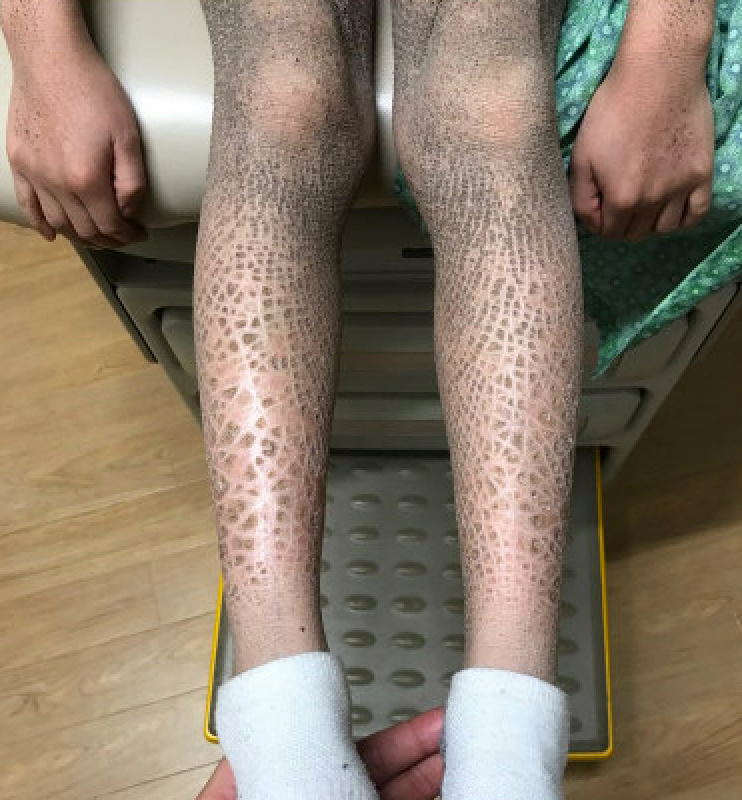
Classically, the scaling in X-linked ichthyosis affects the anterior aspect of the lower extremities while sparing the nails, palms, soles, and flexural areas, such as the popliteal and antecubital fossae.[7] In a series of 30 patients with X-linked ichthyosis, flexural involvement was reported in one-third of cases.[8] In addition, the scalp, preauricular areas, and neck may be affected, resulting in the classical "dirty neck" appearance.
Pruritus is either absent or milder compared to other forms of ichthyosis. Desquamation is typically mild during the summer and exacerbated by dry and cold weather. Additional physical examination findings are listed below.
- Hypohidrosis in 19% of patients.[8]
- Atopic dermatitis in 23% of patients.[8]
- Punctate corneal opacities in 50% of affected males and 25% of female carriers.[9]
- Cryptorchidism in 20% of affected patients.[8]
- Atrial fibrillation or flutter in 10.5% of affected males between the ages of 40 and 69.[7]
- Cognitive and behavioral disorders, especially ADHD, in 30% of affected patients.[7][10]
- Epilepsy.[8]
No significant data suggest adverse effects of the disease on sexual development, testosterone levels, or fertility. [11][12][13] Clinical findings in female carriers of X-linked ichthyosis include corneal opacities and an increased risk of ADHD.
Contiguous Gene Syndromes
Genetic variations may involve genes adjacent to the STS gene, resulting in a group of complex clinical phenotypes. These contiguous gene syndromes manifest as X-linked ichthyosis along with various other syndromes, as listed below.
- Cognitive disabilities.[14]
- Hypogonadotropic hypogonadism and anosmia or Kallmann syndrome (MIM #308700), which occur due to a large deletion proximal to and including the STS gene.[15]
- Chondrodysplasia punctata or stippling near the ends of bones and in cartilage, nasal hypoplasia, developmental delays, and short stature.
- Nystagmus and decreased visual acuity.[16][17][18][19][20]
Evaluation
Some clinical findings that may indicate the possibility of X-linked ichthyosis and should be considered by clinicians are listed below.
- Unusually dry skin during the first weeks of life that evolves into brownish scales with polygonal shapes.
- Labor lasting more than 20 hours or a nonelective cesarean section.
- Male relatives on the maternal side of the family who have scaly skin.
- Low levels of estriol in maternal serum during the second trimester of pregnancy.
Biochemical and genetic analysis form the basis for diagnosing X-linked ichthyosis. Testing steroid synthase activity on cultured fibroblasts provides the most sensitivity, enabling the detection of cases caused by gene deletions and mutations. Furthermore, the chromosomal microarray test of peripheral blood provides diagnostic confirmation. The limitation of chromosomal microarray is its inability to detect point mutations or very small deletions. For patients with suspected X-linked ichthyosis and a negative chromosomal microarray, DNA sequencing is available to identify point mutations and small deletions in the steroid sulfatase gene. The most common technique for DNA sequencing is fluorescence in situ hybridization (FISH).
Chromosomal microarray is a valuable tool to exclude an associated contiguous gene syndrome. For carrier detection, multiplex quantitative fluorescent PCR (QF-PCR) and FISH can detect complete deletion of the STS gene and identify a female carrier. [21][22][23][24] In the prenatal diagnosis of X-linked ichthyosis, experts recommend performing chromosomal microarray to assess the possibility of a contiguous gene syndrome.
Treatment / Management
Currently, no definitive cure for X-linked ichthyosis exists. Although patients with mild disease may not require treatment, those with more severe symptoms may seek intervention due to decreased quality of life and social discomfort. The primary approach to therapy aims to alleviate skin dryness, reduce scales, and enhance skin appearance through daily bathing, regular use of moisturizers and emollients, and the application of keratolytic agents.
Extended showers or daily soaks, combined with the gentle removal of scales using a textured sponge, followed by applying petrolatum or humectant-based moisturizers to damp skin, form the cornerstone of treatment. Humectant-based moisturizers, which include sodium lactate, low-concentration urea, and propylene glycol, are recommended. In addition, the incorporation of baking soda or moisturizing oils into the bath may provide additional benefits.
Older children and adults may incorporate topical keratolytics into their treatment regimen. However, their use is challenging in young children due to the potential for irritation and discomfort. Caution should be exercised when applying topical keratolytics over large body surfaces in children, considering the risk of systemic absorption and associated toxicities. Commonly available keratolytics typically contain 5% to 12% lactic acid, 2% to 10% urea, 5% to 15% glycolic acid, 10% to 25% propylene glycol, and 3% to 6% salicylic acid.
Patients apply keratolytics once or twice daily to the affected areas. The initiation of treatment is gradual to prevent irritation. Initially, patients may apply a small amount to an area once a week and gradually increase the dose. A single keratolytic may not yield adequate results, necessitating combinations such as 5% lactic acid with 10% urea or 2% salicylic acid with 20% urea.
In severe forms of X-linked ichthyosis, patients may benefit from the intermittent use of topical or even systemic retinoids. Limited evidence exists regarding the efficacy of keratolytics, topical, and systemic retinoids for treating mild-to-severe X-linked ichthyosis. Tazarotene, 0.05% gel, has been shown to cause marked clinical improvement in X-linked ichthyosis, whereas oral acitretin has significantly improved scaling and erythema.
Ophthalmology examination is generally unnecessary for affected males as the punctuate corneal opacities are asymptomatic. However, if cryptorchidism is present, a urology consult is warranted, followed by routine monitoring for testicular carcinoma. The association between testicular carcinoma and X-linked ichthyosis is unclear, with only a few existing case reports.[17][18][19]
The thickening of the skin in X-linked ichthyosis can obstruct sweat glands, compromising the patient's ability to regulate body temperature. Skin erythema is an initial indicator of overheating, necessitating access to a cool environment and water for affected patients. In children with contiguous gene syndromes, the management involves the collaboration of various medical specialties tailored to their specific comorbid symptoms.
Differential Diagnosis
The following list comprises the differential diagnoses for X-linked ichthyosis:
- Ichthyosis vulgaris
- Lamellar ichthyosis and congenital ichthyosiform erythroderma
- Atopic dermatitis
- Congenital ichthyosiform erythroderma
- Harlequin ichthyosis
- Epidermolytic ichthyosis
- Sjögren-Larsson syndrome (MIM #270200)
- Anhidrotic ectodermal dysplasia
- Congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome (MIM #308050)
- Conradi-Hünermann-Happle syndrome or Congenital X-linked dominant chondrodysplasia punctata type 2 (MIM #302960)
- Netherton syndrome (MIM #256500)
- Trichothiodystrophy (MIM #601675)
- Multiple sulfatase deficiency (MIM #272200)
- Refsum disease (MIM #266500)
- Keratitis-ichthyosis-deafness syndrome (MIM #148210)
- Neutral lipid storage disease (MIM #275630)
- Ichthyosis prematurity syndrome (MIM #608649)
The following list includes conditions associated with acquired ichthyosis:
- Hodgkin lymphomas
- Anaplastic large-cell lymphoma or lymphomatoid papulosis
- Leiomyosarcoma
- Multiple myeloma
- Cutaneous T-cell lymphoma
- Kaposi sarcoma
- Breast, lung, or bladder carcinoma
- Graft versus host disease
Clinicians can often differentiate acquired ichthyosis from congenital as scaling in acquired disease may also be present on the palms and soles and in skin flexures. The suspected underlying mechanisms are the tumor's secretion of keratinocyte growth factors or an autoimmune response directed against the skin.
Prognosis
Patients without a contiguous gene syndrome generally have an average life expectancy. The quality of life for patients can vary depending on the severity of their symptoms. More severe symptoms may result in increased discomfort and lower self-esteem. Some patients may experience improvement with age, exhibiting milder symptoms in adulthood compared to their childhood.
Clinicians should not overlook the psychological and social impacts of having a visible skin condition. Individuals may benefit from the support of dermatology clinicians, genetic counselors, and psychologists to address the medical and emotional aspects of the condition.[25] With proper management, many individuals with X-linked ichthyosis can lead fulfilling lives. The key to a favorable prognosis lies in early diagnosis, appropriate medical care, and a supportive network of healthcare professionals.
Complications
X-linked ichthyosis primarily affects a patient's skin but can be associated with complications and comorbid conditions. Given below is a detailed overview of potential complications and associated features that may exist in a patient with X-linked ichthyosis:
- Skin infections: The dry, scaly skin in individuals with X-linked ichthyosis can create an environment conducive to bacterial or fungal skin infections. These infections can lead to redness, swelling, pain, and distress.[26]
- Pruritus or itching: Itchy skin is a common and often bothersome symptom of X-linked ichthyosis. Constant scratching can cause breaks in the skin, increasing the risk of infection.
- Atopic dermatitis: Some individuals with X-linked ichthyosis may develop atopic dermatitis in conjunction with their ichthyosis, which can cause additional itching, redness, and skin inflammation.
- Keratosis pilaris: This condition involves the development of small, raised, and often red or white bumps on the skin, primarily on the arms and thighs. Keratosis pilaris can occur in conjunction with X-linked ichthyosis, further complicating skin texture and appearance.[27]
- Retinoid dermatitis: In some cases, retinoids may cause skin irritation and redness as an adverse effect.
- Psychosocial complications: Living with visible skin abnormalities can lead to psychological and social challenges, including low self-esteem, depression, and social isolation.[10]
- Heat intolerance: The thickened, dry skin may make it more difficult for individuals with X-linked ichthyosis to regulate body temperature effectively, causing overheating in hot and humid conditions.
- Prolonged labor and cesarean section: Deficiency of the sulfatase enzyme in the placenta may cause labor initiation or progression failure.
- Cryptorchidism: Cryptorchidism gives rise to additional complications. Children with cryptorchidism are at increased risk of inguinal hernias, testicular torsion, testicular trauma, subfertility, and testicular cancer.
- Testicular germ cell tumors: The actual link between testicular germ cell tumors and X-linked ichthyosis is unclear, as only a few case reports are available in the literature.
- Atrial fibrillation and atrial flutter: The incidence increases in men between the ages of 40 and 69.[7]
- Cognitive and behavioral disorders: Mood disorders, ADHD, anxiety, and depression are more prevalent in affected males and carrier females compared to the general population.[10]
Deterrence and Patient Education
Individuals affected by X-linked ichthyosis should comprehend that although their skin cell production proceeds regularly, the shedding process is slower. To deter complications, adopting meticulous skincare routines is essential. This not only improves the appearance of the skin but also helps prevent skin infections, minimizes itching, and maintains proper body temperature. In addition, avoiding trauma to the skin is crucial for optimal management.
Comprehensive patient education is equally vital. Patients, caregivers, and families should understand the genetic component of X-linked ichthyosis and may benefit from a genetics consultation to better understand the inheritance pattern and impact on relatives. In addition, it is also crucial for patients and caregivers to understand that the condition is lifelong and that a definitive treatment for this condition does not exist. Healthcare professionals must be prepared to address the medical and psychosocial aspects of the disease. By fostering patient understanding, adherence, and informed choices, healthcare providers can empower individuals with X-linked ichthyosis to mitigate complications and improve their quality of life. A valuable online resource for patients, caregivers, partners, friends, and families is The Foundation for Ichthyosis and Related Skin Types (FIRST, https://www.firstskinfoundation.org/).
Enhancing Healthcare Team Outcomes
In addition to thick, dry, scaling skin, patients with X-linked ichthyosis often experience asymptomatic punctate corneal opacities, cryptorchidism, and cognitive or behavioral disorders, especially ADHD. Providing patient-centered care for individuals with X-linked ichthyosis requires a collaborative effort among primary care, psychology, psychiatry, dermatology, ophthalmology, developmental pediatrics, urology, genetics, advanced practice practitioners, and pharmacists. Clinicians must possess the knowledge to recognize, diagnose, and manage X-linked ichthyosis, including understanding the disease's genetic, medical, and psychosocial aspects.
A strategic, evidence-based approach to treatment is necessary to achieve the best possible outcomes while avoiding skin irritation and promoting patient compliance. Efficient interprofessional communication is essential to allow for collaborative decision-making. Care coordination is crucial to foster patient compliance, minimize potential complications, and monitor for psychosocial concerns. Incorporating the principles of skill, strategy, communication, and care coordination into an interprofessional team approach will enhance overall patient care and quality of life while decreasing morbidity in patients with X-linked ichthyosis.