Continuing Education Activity
Atrophoderma of Pasini and Pierini is a rare, cutaneous condition that causes dermal atrophy. Pasini described it in 1923 under the name of ”progressive idiopathic atrophoderma.” Pierini and Vivoli later further described it in 1936, suggesting its possible link to morphea. It presents as single or multiple, sharply demarcated, hyperpigmented, non-indurated patches with no obvious inflammatory signs. This activity describes the presentation, etiology, and pathophysiology of Atrophoderma of Pasini and Pierini and highlights the role of the interprofessional team in its management.
Objectives:
- Identify the etiology Atrophoderma of Pasini and Pierini.
- Review the presentation of Atrophoderma of Pasini and Pierini.
- Outline the treatment and management options available for Atrophoderma of Pasini and Pierini.
- Explain the interprofessional team strategies for improving care coordination and outcomes in patients with atrophoderma of Pasini and Pierini.
Introduction
Atrophoderma of Pasini and Pierini is a rare, cutaneous condition that causes dermal atrophy. Pasini described it in 1923 under the name of ”progressive idiopathic atrophoderma.” Pierini and Vivoli later further described it in 1936, suggesting its possible link to morphea. It presents as single or multiple, sharply demarcated, hyperpigmented, non-indurated patches with no obvious inflammatory signs.
Etiology
The cause of atrophoderma of Pasini and Pierini remains unknown. Some authors have linked it to infection with Borrelia burgdorferi [1] such as Buechner and Rufi who studied the sera of 26 patients with typical lesions. 53% of them had positive immunoglobulin G antibodies against B. burgdorferi, while 14% of control subjects demonstrated positive IgG antibodies. No immunoglobulin M antibodies were found. Some cases of familial atrophoderma of Pasini and Pierini have been described; however, a genetic link has not been confirmed. [1]
A neurogenic cause has been suggested because of the apparent zosteriform distribution of lesions in some cases, but definitive evidence is lacking. [2] There is growing evidence to consider the atrophoderma of Pasini and Pierini as a manifestation of the spectrum of localized scleroderma. [3], [4]
Various classification schemes have been proposed. The first was suggested in 1995 by Paterson and classifies morphea into five groups: plaque, generalized, bullous, linear, and deep. Atrophoderma of Pasini and Pierini was included within the spectrum of plaque-type morphea along with Morphea en plaque, guttate morphea, keloidal morphea, and lichen sclerosus et atrophicus. Generalized morphea was defined as involving two or more body areas. Bullous or linear morphea included linear morphea of the extremities, morphea en coup de sabre, and progressive hemiatrophy. Deep morphea in this system included morphea profunda, subcutaneous morphea, eosinophilic fasciitis, and pansclerotic morphea.
Cases of "burnt-like" presentation of morphea may be atrophic and histologically similar to atrophoderma of Pasini and Pierini. Both morphea plaques and atrophoderma of Pasini and Pierini lesions may occur in the same patient, with atrophic patches appearing in the previously normal skin. In one case report [5] the authors described a patient with typical atrophoderma of Pasini and Pierini who developed progressive systemic sclerosis.
Epidemiology
Atrophoderma of Pasini and Pierini is a rare disease. Less than 100 cases have been reported in the literature. It is usually seen in the second or third decade of life. However, the onset of the disease has also been described in younger and elderly patients. Cases of congenital atrophoderma have also been reported. [2] Atrophoderma of Pasini and Pierini has a marked female preponderance, with a female to male patient ratio of 6:1. The condition is more frequently reported in white Europeans.
Histopathology
Microscopic findings in the atrophoderma of Pasini and Pierini are not striking. In many cases, no alterations can be observed in the epidermis, but it may show slight atrophy with flattened rete ridges. The most common finding is a quantitative decrease in the thickness of the affected dermis. Collagen changes can include atrophy, sclerosis, fragmentation and hyalinization [6] Elastic fiber changes may be variable; reduction and fragmentation similar to that of anetoderma cases have been reported [5] Dilatation of superficial blood vessels is noted in some reports associated with a sparse, perivascular round-cell inflammatory infiltrate. The sweat glands, pilosebaceous units, and appendages appear normal.
History and Physical
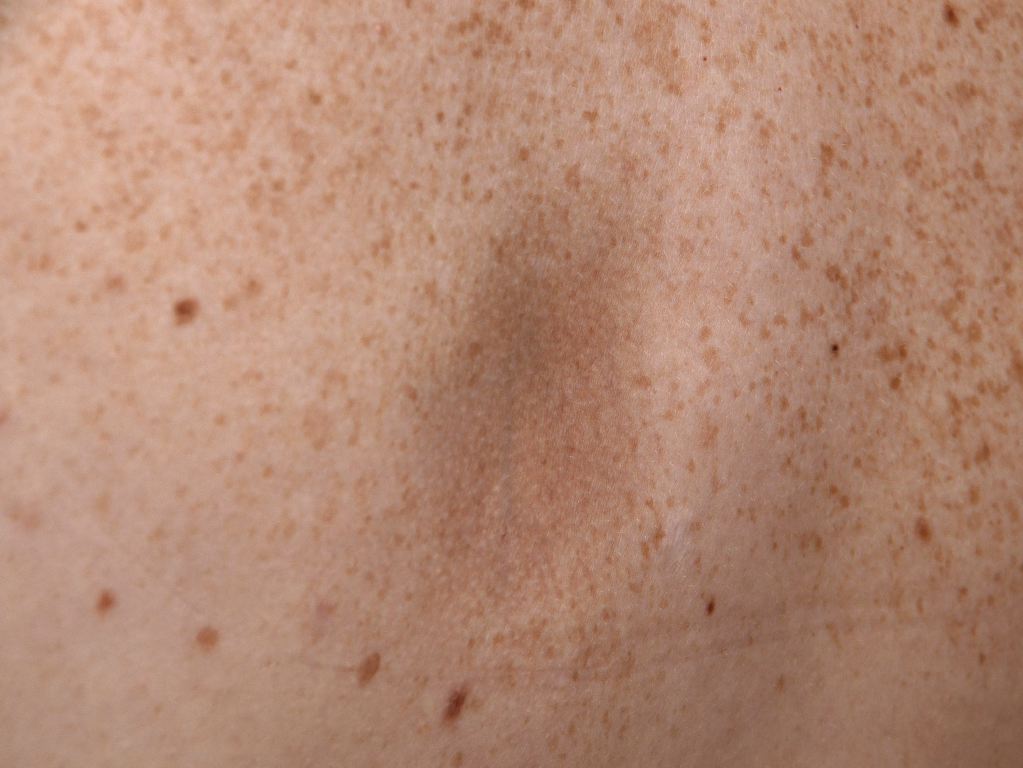
From a clinical perspective, the condition is asymptomatic and progresses insidiously over time. The lesions first appear on the trunk (commonly on the back) and progress to involve chest, arms, and abdomen. It may cause pain, pruritus, or even paraesthesia.
It is characterized by single or multiple [3] irregularly round patches, varying in size from a few millimeters to several centimeters. These patches are slightly depressed (below the level of surrounding skin) with a sharp edge, giving a "cliff-drop" border appearance. An erythematous or "lilac-ring" border, as seen in morphea, does not occur in atrophoderma of Pasini and Pierini. The lesions are usually hyperpigmented, but Saleh et al. [5] have shown in a retrospective study of 16 Lebanese patients that the lesions could be hypopigmented (56%) and skin-colored (25%). Therefore, they may be hypopigmented, as well.
Lack of features of inflammation is a characteristic finding. The lesions do not exhibit sclerosis or induration on palpation. Surrounding skin appears normal and does not show any changes. The lesions are often bilateral; however, unilateral zosteriform or blaschkoid presentation has been documented. [3]
Evaluation
An enzyme-linked immunosorbent assay should be performed to detect anti–B. burgdorferi antibodies.
The thickness of the lesion and surrounding skin may be measured with magnetic resonance imaging or 13-MHz B-mode ultrasonography, to document dermal atrophy of lesional area. However, it is often not needed.
Histopathological changes are minimal and show the reduced volume of dermal tissue as compared to normal skin. Some notable findings include varying degrees of homogenization and clumping of collagen in the mid- and reticular dermis, interstitial edema, and a mild perivascular infiltrate consisting of lymphocytes and histiocytes. The sweat glands, pilosebaceous units, and appendages appear normal. The overlying epidermis is usually normal but may show slight atrophy with flattened rete ridges.
Treatment / Management
Unfortunately, no treatment is consistently effective for atrophoderma of Pasini and Pierini.
Patients with a positive B. burgdorferi antibody titer have shown some response with tetracyclines. Doxycycline, in a dose of 200 mg/day, is reported to prevent the appearance of new lesions. However, the results of treatment with antibiotics in a patient with positive B. burgdorferi are inconclusive. In retrospective sera of 25 patients treated with either oral penicillin (2 million IU per day) or oral tetracycline (500 mg three times per day ) for 2 to 3 weeks showed clinical improvement in 20 patients. The same study demonstrated no progressive disease in four of six patients who did not receive treatment [7]
The therapeutical approach also includes topical corticosteroids and antimalarials, including hydroxychloroquine, which seems to show good response in cases associated with lupus. Furthermore, topical treatments using calcineurin inhibitors were also reported, although with variable responses [8].
Treatment with a Q-switched Alexandrite laser has also been reported to result in the clinical improvement of hyperpigmented lesions. [9]
Differential Diagnosis
- Atrophoderma of moulin (LAM)
- Postinflammatory hyperpigmentation
Prognosis
The disease tends to be progressive over 10 to 20 years. Eventually, no further progression occurs, yet existing lesions characteristically do not involute. No definite progression to systemic sclerosis had been observed until Bisaccia et al. [10] in 1982, which reported the case of a 42-year-old woman with typical lesions of the atrophoderma of Pasini and Pierini on the back. The patient developed progressive systemic sclerosis with Raynaud phenomenon, sclerodactyly, and pulmonary fibrosis.
Enhancing Healthcare Team Outcomes
Atrophoderma of Pasini and Pierini is a rare disease that most likely will not be encountered by the primary care provider or nurse practitioner. However, when faced with unknown skin lesions, the best recommendation is to refer the patient to a dermatologist for a definitive diagnosis and then work as a team to provide coordinated follow up. Atrophoderma of Pasini and Pierini has a marked female preponderance, with a female to male patient ratio of 6:1. The condition is more frequently reported in white Europeans. The disorder may progress in some people, but little is known about its long term course.
Due to the challenge of dealing with a rare disease, it should be managed with an interprofessional team. The pharmacist should assist with medication reconciliation, and treatment options as the patient may need topical steroids and antimalarials as well as antibiotics. The pharmacist should also assist with patient education and assist in monitoring compliance. The clinician should have the support of a dermatology specialty nurse to assist with patient and family education. Further, the nurse should make sure regular monitoring is attained, and close follow-up visits are arranged. Any concerning issues should be reported to the interprofessional team. The best outcomes will be achieved with a coordinated approach to treatment, education, and monitoring. [Level 5]