Continuing Education Activity
Apert syndrome is an autosomal dominant inherited craniosynostosis syndrome. Males and females are equally affected. The incidence of the disease significantly increases with paternal age and is felt to provide a selective advantage within the male spermatogonial cells. This activity describes the evaluation, diagnosis, and management of Apert syndrome and highlights the role of team-based interprofessional care for affected patients.
Objectives:
- Describe the pathophysiology of Apert syndrome.
- Review the physical features of Apert syndrome.
- Outline the treatment and management options available for Apert syndrome.
- Explain interprofessional team strategies for improving care coordination and communication to advance the treatment of Apert syndrome and improve outcomes.
Introduction
Apert syndrome is another genetically inherited syndrome characterized by craniosynostosis (premature fusion of coronal sutures) resulting in skull and facial deformities and syndactyly. The syndrome was first described in 1906 by French physician Eugene Apert when he described nine people with similar facial and extremity characteristics.[1]
Etiology
Apert syndrome is an autosomal dominant inherited craniosynostosis syndrome. It is due to gain-of-function missense mutations of fibroblast growth factor receptor (FGFR2)-2 on chromosome 10q.[2]
Epidemiology
Apert syndrome is a rare disease and is estimated to occur in 1 in 65,000 to 200,000 births depending on the study cited.[3] Males and females are equally affected. The incidence of the disease significantly increases with paternal age and is felt to provide a selective advantage within the male spermatogonial cells.[4] The syndrome has complete penetrance but variable expressivity resulting in phenotypically unaffected to severe deformities within the same family.
Pathophysiology
Two-thirds of cases of Apert syndrome are due to a specific cytosine to guanine mutation at position 755 of the fibroblast growth factor receptor 2 (FGFR2) gene resulting in a serine to tryptophan amino acid change on the paternally derived allele.[4][5] The incidence of the disease increases with the age of the father.[4] Unfortunately, there are only hypotheses as to why both the extremities and cranial sutures are affected, and some data from a single mouse model. In mice, the FGFR2 receptor loses its specificity and can bind other fibroblast growth factors, thereby suppressing apoptosis of osteoblasts resulting in syndactyly and craniosynostosis. The underlying mechanism is still not clear even in this mouse model, but it is linked to a specific FGF.[6]
History and Physical
The family history of patients suspected of having Apert syndrome is crucial due to its autosomal dominant inheritance. A lack of family history does not rule out the diagnosis due to the possibility of de novo mutations; however, a positive family history makes the diagnosis much more likely.
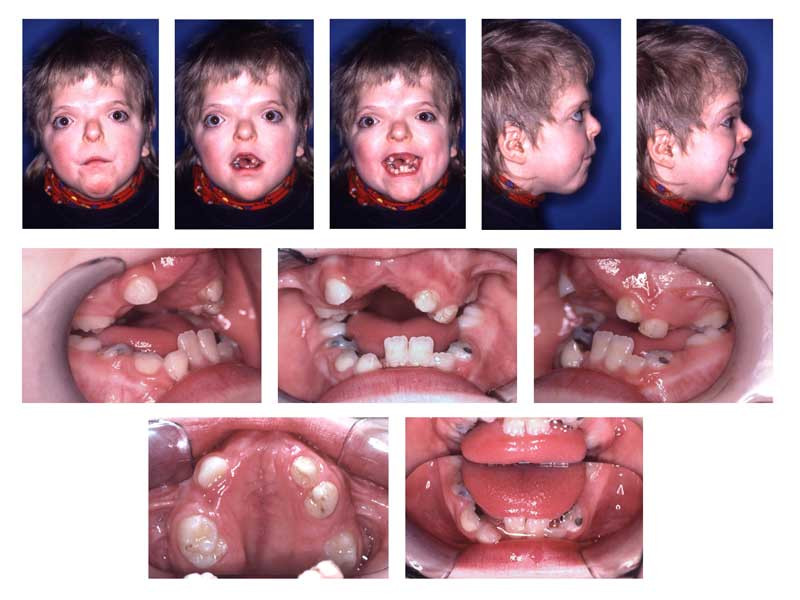
Patients with Apert syndrome have craniosynostosis, midface hypoplasia, and symmetric syndactyly of the hands and feet. The craniosynostosis is more severe than that found in Crouzon syndrome, and the additional finding of syndactyly helps confirm the diagnosis between multiple, similar syndromes in regards to their phenotype. However, features of hypertelorism (wide-set eyes), proptosis (bulging eyes), and down-slanting palpebral fissures are facial features found in several of the craniosynostoses that cannot be used to differentiate the syndromes but are helpful.
In regards to the hand, a short, radially deviated thumb, complex syndactyly of the index, long, and ring finger, syndactyly of the fourth webspace, and symphalangism (congenital stiffness of the fingers due to failure of the bone to fully separate as typically happens during fetal growth) are characteristic findings of the hand. There are three specific subtypes of hand findings in Apert syndrome based on the overall shape of the hand. They are spade (side-to-side fusion with flat palm), mitten (fusion of fingers resulting in concave palm), and rosebud (tight fusion of all digits). There are other craniofacial deformities found in Apert syndrome and other craniosynostosis, such as acrocephaly (cone-shaped calvarium), proptosis, prominent forehead, hypertelorism, down slanting palpebral fissures, and a flattened nasal bridge. Oral findings included dental crowding, high-arched palate, narrow palate, and pseudo-clefts. Internal organs and other skeletal anomalies, such as cervical fusions, can be seen. Mild to moderate intellectual disability is also possible.
Evaluation
The evaluation for Apert syndrome in the setting of known family history is a clinical one, as the characteristic physical examination findings confirm the diagnosis. In cases where the clinical presentation is not clear and no family history to support the diagnosis, additional tests such as advanced imaging techniques can help. Magnetic resonance imaging (MRI) and computed tomographic (CT) imaging of the brain are used to detect craniosynostosis or other skeletal abnormalities (peri sutural sclerosis, reduced serration, and bony bridging and/or the absence of the suture altogether). These same imaging techniques can be helpful in detecting complications related to the syndrome, such as increased intracranial pressure.
Much like that discussed with Crouzon syndrome, in which there is an unclear diagnosis or the syndrome has atypical features, genetic and molecular testing can be pursued. Unfortunately, the underlying mechanism of multiple craniosynostosis syndromes is related to FGFR mutations and abnormal signaling. Prenatal genetic testing, MRI, and ultrasounds can be utilized to confirm the diagnosis before the birth of the child.[7] The use of amniocentesis and/or chorionic villus sampling can be performed, but the combination of safer imaging techniques will likely render the higher-risk procedures obsolete except in the most difficult cases.
As described above, the history, physical, and imaging findings are used to confirm the specific craniosynostosis but can be difficult due to significant overlap amongst the syndromes (Pfeiffer, Apert, Saether-Chotzen, Carpenter, and Jackson-Weiss syndromes).
Treatment / Management
Like other craniosynostoses, management is a team-based approach requiring multiple subspecialists such as pediatricians, neurosurgeons, plastic surgeons, craniofacial surgeons, ophthalmologists, and dentists. Surgery is required to prevent complete coronal suture closure and protect brain development.
A detailed description of the surgical management of Apert syndrome can be found in the guidelines from the “Working Group on Craniosynostosis.[8]” Much like Crouzon syndrome, earlier surgical decisions (before the age of 1) are felt to provide better long-term outcomes.[9] Unfortunately, this is based on anecdotal evidence and not a randomized, controlled trial. Similarly, there is no standard of care for the treatment of syndactyly, but multiple revisions are likely as the child grows.
Long-term follow-up is essential to reduce the risk of developing complications related to craniosynostosis, such as strabismus, sleep apnea, and elevated intracranial pressure. Unfortunately, these issues are not completely resolved with surgical correction of the facial and cranial defects, as 54% of patients had vision loss in at least one eye related to amblyopia that developed after craniofacial surgery for Apert syndrome in one retrospective study from Australia. Fortunately, in this same study, the incidence of optic atrophy was low at 5%, presumably due to the widespread adoption of early craniofacial surgery for craniosynostosis syndromes.[10] The incidence of strabismus is extremely common as well, with two-thirds of patients developing the entity at some point.[11] Severe to profound hearing loss is also much more common in syndromic craniosynostoses than in nonsyndromic variants.[12] Thus, a team-based approach with multiple subspecialists is involved to monitor for the development of vision and life-threatening complications related to Apert and to make difficult decisions and if and when to perform surgery.
While only available in laboratories at this point, chemical inhibitors of the FGFR signaling pathway restore normal FGFR signaling and rescue the associated skeletal defects.
Differential Diagnosis
The differential diagnoses of Apert syndrome include:
- Achondroplasia
- Antley-Bixler syndrome
- Beare-Stevenson syndrome
- Conditions arising due to mutations of the fibroblast growth factor receptors
- Crouzon syndrome
- Cutis gyrata
- Pfeiffer syndrome
- Thanatophoric dysplasia
Prognosis
The prognosis depends on when surgery was performed and what pathologies required surgical intervention.
Complications
The main complications likely to occur in patients with Apert syndrome include:
- Increased intracranial pressure that can cause papilledema and cognitive impairment
- Exposure keratopathy and corneal scarring
- Respiratory complications
- Spinal cord injury and neurologic deficits in patients with cervical spine anomalies
- Aspiration pneumonia and further chronic lung disease
Consultations
- Ophthalmology (pediatric, oculoplastics)
- Plastic surgery
- Maxillofacial surgery
- Neurosurgery
- Otorhinolaryngology
Deterrence and Patient Education
Apert syndrome is having an autosomal dominant inheritance, and advanced paternal age is found to be associated with the de novo occurrence of Apert syndrome. There is a 50% chance of the genetic trait being passed to each child. If a pathologic variant person is present in the family, prenatal testing for pregnancies at increased risk should be ideally performed.
Enhancing Healthcare Team Outcomes
Like other craniosynostoses, management is a team-based approach requiring multiple subspecialists such as pediatricians, neurosurgeons, plastic surgeons, craniofacial surgeons, ophthalmologists, and dentists. Surgery is required to prevent complete coronal suture closure and protect brain development.
Long-term follow-up is essential to reduce the risk of developing complications related to craniosynostosis, such as strabismus, sleep apnea, and elevated intracranial pressure. Unfortunately, these issues are not completely resolved with surgical correction of the facial and cranial defects, as 54% of patients had vision loss in at least one eye related to amblyopia that developed after craniofacial surgery for Apert syndrome in one retrospective study from Australia. Fortunately, in this same study, the incidence of optic atrophy was low at 5%, presumably due to the widespread adoption of early craniofacial surgery for craniosynostosis syndromes.[10] The incidence of strabismus is extremely common as well, with two-thirds of patients developing the entity at some point.[11] Severe to profound hearing loss is also much more common in syndromic craniosynostoses than in nonsyndromic variants.[12] Thus, a team-based approach with multiple subspecialists is involved to monitor for the development of vision and life-threatening complications related to Apert and to make difficult decisions and if and when to perform surgery.