Introduction
Normal facial development enables individuals to perform critical auditory, visual, breathing, masticatory, expressive, and vocal functions. Facial primordia appears near the mouth's future site in the 4th week of development. Henceforth, facial development becomes a complex process involving the formation of various primordial tissues.
Many congenital facial anomalies have genetic causes, though some arise from preventable teratogen exposures. Developmental facial abnormalities can be disfiguring and devastating. The treatment of congenital facial disorders is often delayed until after birth because they are well-tolerated in utero.[1]
Development
Facial Primordia
The incipient stomodeum or stomatodeum helps produce the mouth and can be seen in the embryo as early as 3 weeks post-fertilization. This structure is surrounded by the developing facial primordia. The stomodeum appears between the enlarging areas of the incipient heart and brain and is later closed off from the cranial foregut by an ectodermal proliferation called the "oropharyngeal membrane." Facial development involves all primary embryonic tissues: ectoderm, mesoderm, and endoderm. This highly coordinated process begins between weeks 4 and 8 post-fertilization and is induced by organizing centers.[2]
Early during facial development, 5 facial primordia appear as processes or prominences surrounding the stomodeum:
- Frontonasal process - arises from mesenchyme produced by the neural crest cells near the forebrain and ventral midbrain. The frontal region of the frontonasal process produces the forehead, while the nasal portion produces the nose.
- Maxillary processes - paired structures arising from the 1st pharyngeal arches superior to the mandibular processes. Mesoderm and neural crest cells migrate into the 1st pharyngeal arches to generate maxillary mesenchyme during the 4th week of development. The maxillary processes will later produce a pair of palatal processes. the maxilla or upper jaw, the zygoma or cheekbone, and the squamous portion of the temporal bone.
- Mandibular processes - paired structures derived from the 1st pharyngeal arches. The 1st pharyngeal arches are also known as the mandibular arches. These processes will produce the mandible or lower jaw.
These primordia fuse to form the face.
The nasal placodes are 2 surface ectodermal thickenings appearing at the end of the 4th week on the inferolateral portion of the frontonasal process.[3] The olfactory epithelium arises from the nasal placodes and neural crest cells. In the 5th week of development, mesenchymal proliferations beside the nasal placodes produce 4 swellings. These protrusions arise from the frontonasal process and include 2 medial and 2 lateral nasal processes that are medial and lateral to the nasal placodes, respectively. Once the 4 swellings form, the nasal placodes become shallow basins called "nasal pits," which then become the nasal cavities and the ventral portion of the nostrils, also known as the nares.
The paired maxillary processes grow larger medially toward the nasal and contralateral maxillary processes. The maxillary processes then fuse with the lateral nasal processes to form the nasolacrimal groove, the site where the cheek and lateral nasal territory merge. A rodlike ectodermal development forms at the nasolacrimal groove's base, producing an epithelial cord that canalizes to form both the nasolacrimal duct and lacrimal sac.
The maxillary processes expand midline to merge with the medial nasal processes. Fusion with the medial nasal processes results in mesenchymal commingling in the intermaxillary territory, producing a single, continuous maxilla and the upper lip and palate. The premaxilla and gingival lining, primary palate, and deep median portion of the labium superiorus (upper lip) arise from the intermaxillary segment. The lateral portion of the labium superiorus, most of the maxilla, and the secondary palate derive from the maxillary processes. The maxillary process outgrowths cover the inferior aspect of the medial nasal processes to form the philtrum of the upper lip. An orofacial cleft occurs when the processes fail to fuse properly.
Other relevant events happen during this period of facial development. In the 5th week, the oropharyngeal membrane degenerates, producing an opening between the cranial foregut and the extraembryonic environment. Myoblasts from the 1st pair of pharyngeal arches produce the mastication muscles, while myoblasts from the 2nd pair of pharyngeal arches produce the facial expression muscles. The eyes begin to move ventrally from their initial lateral positions as the rest of the head grows and develops. The external ears appear in the cervical region and then rise to the head's lateral aspect. By the end of developmental week 7, the embryo has facial features with a typical human-like appearance.
Nasal Cavities
Nasal cavity formation revolves around nasal placode development. The paired medial and lateral nasal processes are tissue elevations that form around the nasal placodes and turn them into shallow depressions called "nasal pits." Meanwhile, the nasal sacs expand dorsally toward the forebrain. The nasal sacs are nasal cavity primordia separated from the oral cavity by a membrane, the oronasal membrane. The membrane eventually ruptures, creating an opening between the nasal and oral cavities.
The choanae are areas posterior to the palate, connecting the oral and nasal cavities. The conchae or turbinates develop from the lateral wall of the nasal cavities. The specialized olfactory epithelium forms from the superior aspect of the nasal cavities. The paranasal sinuses—the air-filled extensions of the nasal cavity—appear late in fetal development as nasal cavity diverticula.
Palate
The palate lies between the nasal and oral cavities and arises from the primary and secondary palates. By the 6th week, the intermaxillary segment develops from the fusion of the paired medial nasal processes and maxillary processes. This fusion gives rise to the primary palate. The oronasal membrane develops posteriorly as the barrier between the nasal and oral epithelia becomes thinner.[3] The membrane later ruptures and forms the primitive choanae that connect the oral cavity to the nasal cavity. The primary palate will give rise to the premaxilla, a triangular region occupying the anterior one-third of the palate. The premaxilla bears the incisive foramen and 4 upper incisors.
The secondary palate forms the rest of the hard palate and all of the soft palate. This region develops during the 7th and 8th weeks post-fertilization. The secondary palate forms from 2 palatal shelves—the lateral palatal processes—that are medial outgrowths of the maxillary processes. These processes grow inferiorly and parallel to the tongue. By the 8th week's end, the lateral palatal processes fuse and, with the primary palate, form the definitive palate. During this time, the nasal septum grows, separating the left and right nasal passages. The septum's inferior portion will merge with the definitive palate. Disruption of these developmental processes may result in the formation of a cleft palate.
Pharyngeal Apparatus
The pharyngeal or branchial apparatus first appears during the 4th week of development and is critical in forming head and neck structures. The pharyngeal apparatus includes the pharyngeal arches, grooves or clefts, and pouches, all of which are comprised of mesenchymal tissue lined externally by ectoderm and internally by endoderm.
The pharyngeal grooves arise from the approximation of ectodermal tissue between consecutive pharyngeal arches, while the pharyngeal pouches form from the approximation of endodermal tissue between consecutive pharyngeal arches. The derivatives of the pharyngeal apparatus crucial to facial development are discussed below.
Pharyngeal groove or cleft
The pharyngeal grooves originate from the ectoderm. The 1st pharyngeal groove produces the external auditory meatus and part of the middle ear. The rest of the pharyngeal grooves form the cervical sinus, which typically degenerates during development. Pharyngeal membranes form on the floor of the pharyngeal grooves. The pharyngeal membrane of the 1st pharyngeal groove gives rise to the tympanic membrane.
Pharyngeal pouch
The pharyngeal pouches are lined by endodermally derived epithelium. The 1st pouch forms the auditory tube, tympanic cavity, and mastoid antrum. The 2nd pouch contributes to the palatine tonsillar tissues. The 3rd pouch forms the inferior parathyroid gland and thymus. The 4th pouch contributes to the superior parathyroid gland.
Pharyngeal arch
The pharyngeal arches originate from the mesoderm. The table below summarizes the adult derivatives of the pharyngeal arches.[4]
| Pharyngeal Arch |
Adult Derivatives |
| 1st pharyngeal arch |
- Maxillary (CN V2) and mandibular (CN V3) branches of the trigeminal nerve (CN V)
- Mandible
- Incus
- Malleus
- Muscles of mastication
- Tensor veli palatini
- Tensor tympani
- Mylohyoid muscle
- Anterior belly of the digastric muscle
- Maxillary artery
- Sphenomandibular ligament
- Meckel cartilage
|
| 2nd pharyngeal arch |
- Facial nerve (CN VII)
- Stapes
- Stapedius
- Styloid process
- Stylohyoid ligament and muscle
- Superior body of the hyoid
- Lesser horn of the hyoid
- Muscles of facial expression
- Posterior belly of the digastric.
|
| 3rd pharyngeal arch |
- Glossopharyngeal nerve (CN IX)
- Inferior body of the hyoid
- Greater horn of the hyoid
- Stylopharyngeus muscle
|
| 4th pharyngeal arch |
- Superior laryngeal branch of the vagus nerve (CN X)
- Thyroid, corniculate, and cuneiform cartilages
- Levator veli palatini
- Pharyngeal muscles
- Extrinsic laryngeal muscles
|
| 6th pharyngeal arch |
- Recurrent branch of the vagus nerve (CN X)
- Arytenoid cartilage
- Intrinsic laryngeal muscles
|
Cellular
Cranial neural crest cells and the endoderm, mesoderm, and ectoderm are critical to facial tissue development. The cranial neural crest cells derive from the ectodermal leaflet from the dorsal midline region. These cells migrate toward the pharyngeal arches and the frontonasal process to contribute to the skull and upper cervical tract tissues. Various signaling pathways play critical roles in head and neck development, including bone morphogenic proteins {BMPs), fibroblast growth factor (FGF), sonic hedgehog (SHH), and wingless-related integration site (WNT).
The ectodermal leaflet at week 4 overlies the stomodeum. Thus the ectoderm is in direct contact with the endodermal leaflet following oropharyngeal membrane formation. During the 5th week, the ectoderm fuses with the mesoderm to start forming the nasal processes. Between the 4th and 5th weeks, the cells of the 3 layers meet to form the facial structures.
Biochemical
Mechanical-chemical waves impact tissue morphogenesis and overall development, as they play a vital role in intercellular communication. The signals produced direct tissue morphogenesis. Chemical reactions at the level of the microtubule organizing center (MTOC) initiate the mechanical-chemical waves.
Molecular Level
The ectodermal placodes, which give rise to future sense organs and cranial ganglia, develop different molecular responses. In the placodes' anterior aspect, coding molecules express paired box protein (Pax) and the homeobox proteins Six3 (SIX3) and Otx2 (OTX2). Other molecules are present in the placodes' posterior portion, such as Iroquois-class homeodomain protein (IRX-1) and the homeobox protein Gbx2 (GBX-2). These proteins help regulate differentiation. For example, Pax6 is more concentrated in the olfactory and lens developmental areas, whereas Pax3 and Pax2/8 help form the trigeminal ganglia and auditory areas.
Function
The facial musculature arises from prechordal mesenchyme and the unsegmented paraxial mesoderm. The prechordal mesenchyme derives from the prechordal plate, which lies anterior to the notochord. The facial musculature serves multiple functions, including feeding, relaxing, breathing, mastication, and facial expressions.
Pathophysiology
Facial developmental anomalies may be due to genetic or environmental causes. Pregnancy-related etiologies include fetal alcohol syndrome, uterine growth restriction, oligohydramnios, and maternal infections.[5] Genetic conditions that can cause abnormal facial development include Pierre-Robin, Treacher-Collins, Fragile X, Down, and DiGeorge syndromes.[6][7]
An orofacial cleft may likewise be due to genetics, environmental factors, or both. This condition is known to run in families. Several genes are implicated, including the cleft lip and palate transmembrane protein 1 (CLPTM1), poliovirus receptor-related 1 (PVRL1), and γ-aminobutyric acid receptor subunit β-3 (GABRB3) genes.[8][9]
Cleft palates may occur as part of genetic conditions like Treacher-Collins, Stickler, and Loeys-Dietz syndromes.[10][11] Environmental factors found to cause palatal clefts include fetal hypoxia from maternal smoking, alcohol abuse, maternal anticonvulsant therapy, and retinoid intake.
Holoprosencephaly (HPE) occurs due to failed cerebral hemisphere separation and is associated with forebrain and facial midline defects.[12] The condition may result from SHH signaling dysfunction or altered bone morphogenic proteins (BMPs).
Clinical Significance
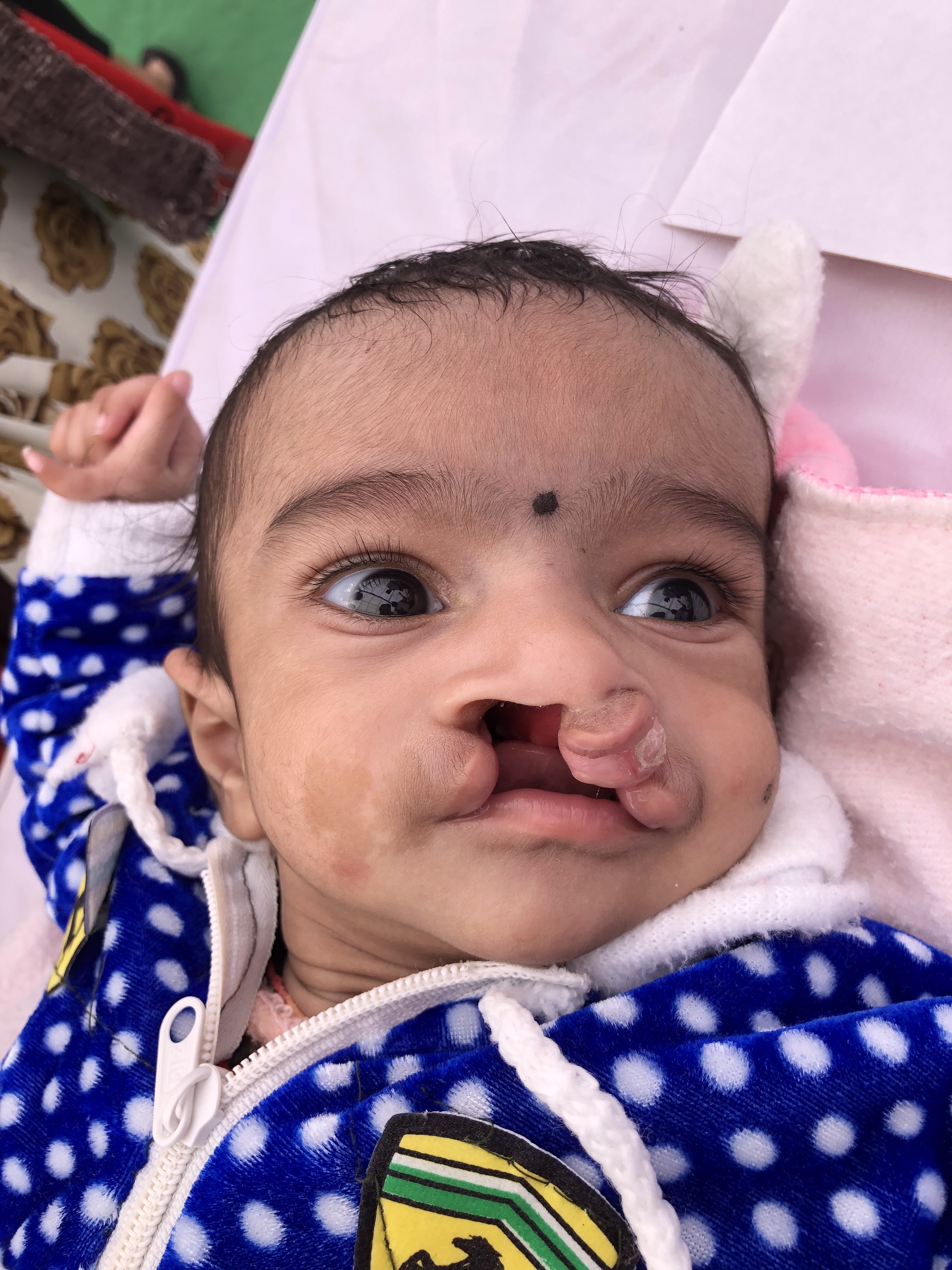
Clinically, one of the most common facial anomalies is an oral cleft, which can be in the form of a cleft lip, cleft palate, or a combination of both (see Image. Oral Cleft). The World Health Organization reports that oral cleft occurs in about 1 in every 700 live births worldwide. This condition is the second most common congenital anomaly in the United States, affecting 1 in 940 births and producing 4437 cases every year. The diagnosis is made clinically at birth but can also be done by ultrasonography in utero. Orofacial cleft complications may include feeding, speech, and cognitive difficulties depending on severity. The definitive management for cleft lip or palate is surgery, although the technique depends on the type and severity of the condition.
Choanal atresia is a less common but potentially fatal congenital anomaly. The oronasal membrane (choana) normally recanalizes during fetal development. However, failure of this process can result in nasal obstruction by abnormal tissue. Choanal atresia may occur as part of CHARGE syndrome (see Image. CHARGE Syndrome). "CHARGE" stands for Coloboma, Heart disease, Atresia choanae, Retarded growth and development, Genital underdevelopment, and Ear anomalies.[13]
The presentation of choanal atresia differs based on severity. Unilateral choanal atresia may go undetected because the newborn can breathe with the normal nostril. However, bilateral blockage can be life-threatening. The neonate may present with cyanosis when feeding, as they cannot use their mouths to compensate for the lack of nasal breathing. The cyanosis generally resolves when the baby starts to cry. The inability to pass a nasogastric tube can be diagnostic and may be confirmed by a computed tomography (CT) scan. The definitive treatment for choana atresia is surgery.
A complete newborn physical examination is crucial. Facial anomalies should prompt clinicians to search for other congenital conditions. Head and neck anomalies often occur as a part of developmental syndromes.