Introduction
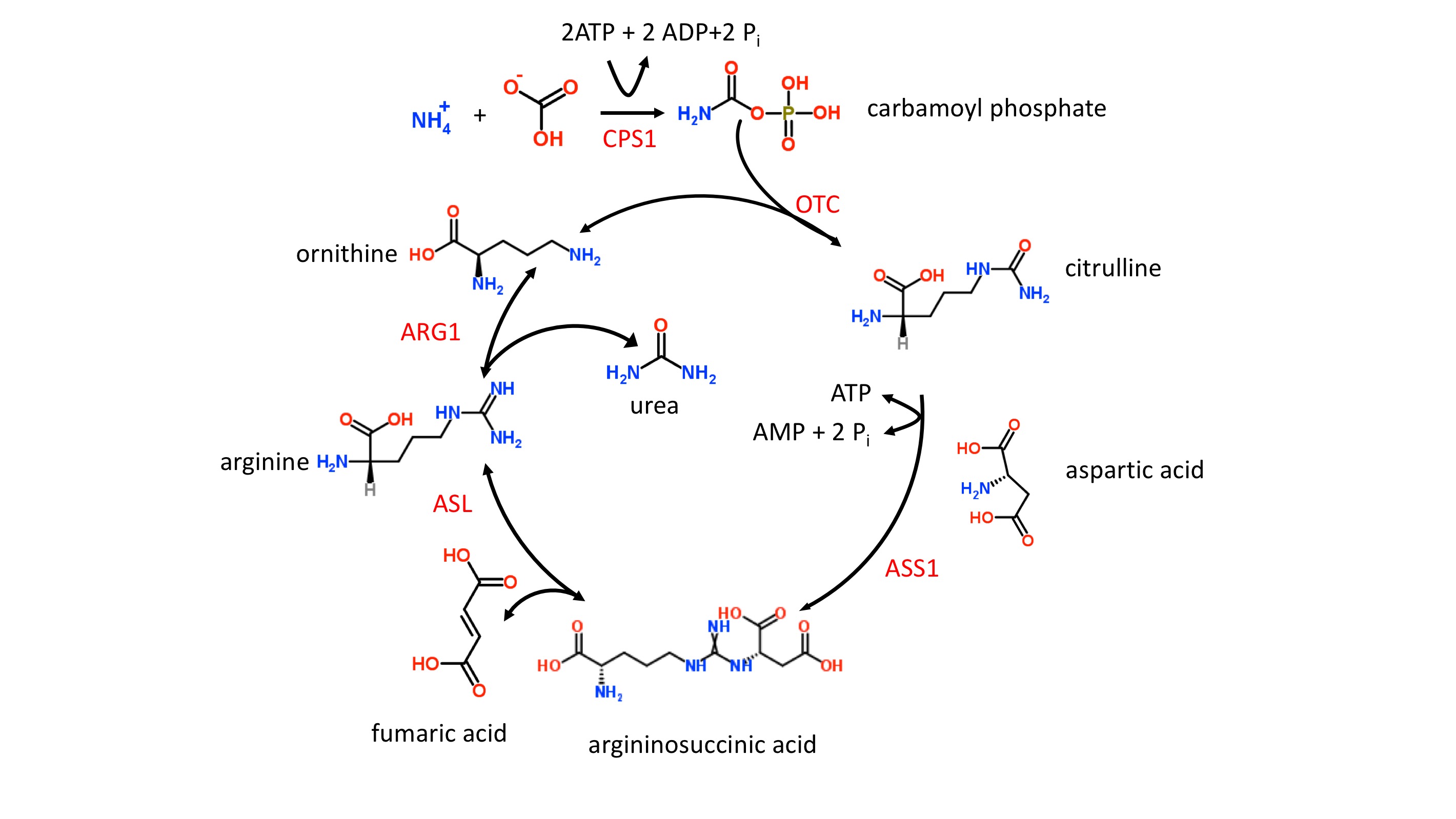
The liver catabolizes toxic ammonia into urea, as shown in figure 1. This energy-dependent process occurs only within the liver’s mitochondria and cytoplasm. When this process is not working efficiently, toxic ammonia (NH3) accumulates within the body and may elicit clinical manifestations such as lethargy, slurred speech, cerebral edema, and asterixis. Here, we will review the underlying physiology of the urea cycle.[1][2][3]
Cellular Level
The urea cycle begins in the mitochondria of hepatocytes and ends in the cytoplasm. The following reactions will be shown schematically below each described step. Note that the enzyme responsible for each respective step will not be shown but can be found in the preceding text.
Step 1
- The first step, which is also rate-limiting, involves the conversion of CO and ammonia into carbamoyl phosphate via the enzyme carbamoyl phosphate synthetase I (CPS I). Ammonia is the source of the first amine group of urea. What is unique about this step is that CPS I requires an obligate activator, N-acetyl-glutamate (NAG). NAG arises from glutamate + acetyl-CoA via the enzyme NAG synthase, which can be upregulated by arginine. Of note, some sources may use NH and HCO+ as the initial reactants, but these are equivalent to CO+ HO + NH.
- CO+ NH+ 2ATP produces carbamoyl phosphate +2ADP + P
- Synthesis of NAG: glutamate + acetyl-CoA NAG
Step 2
- Carbamoyl phosphate and ornithine combine to form citrulline via ornithine transcarbamoylase (OTC). Citrulline is then transported from hepatocyte mitochondria into the cytoplasm by ornithine translocase.
- Carbamoyl phosphate + ornithine citrulline
- Citrulline in mitochondria citrulline in the cytoplasm
Step 3
- Citrulline reacts with aspartate to form argininosuccinate. This reaction is carried out by the enzyme argininosuccinate synthetase, which requires ATP. Aspartate is the source of the second amine group on urea. Recall that aspartate results from the transamination of oxaloacetate and glutamate via aspartate transaminase, which requires vitamin B.
- Citrulline + aspartate + ATP argininosuccinate
- Oxaloacetate + glutamate à aspartate + alpha-ketoglutarate
Step 4
- Argininosuccinate is converted into arginine via argininosuccinate lyase. This reaction also gives off fumarate, which is involved in the mitochondrial generation of NADH in the TCA cycle, as well as tyrosine catabolism.
- Argininosuccinate à arginine + fumarate
Step 5
- Arginine undergoes hydrolysis via arginase to form urea and ornithine. Take a moment to review step 2. Note that the regeneration of ornithine in step 5 is involved in step 2.
- Arginine + HO urea + ornithine
Organ Systems Involved
The liver is the only site where urea is synthesized and ultimately excreted by the kidneys.
Function
The urea cycle is the body’s way of converting toxic ammonia into urea. Ammonia originates from protein catabolism whether that is secondary to a high-protein diet, deaminations, or during the period of prolonged starvation. Ammonia is also naturally produced by gut flora. In muscle and peripheral tissues, glutamate is the amino acid that accepts free ammonia, which results in the formation of glutamine. Glutamine is then exported from muscle and peripheral tissues and utilized by the liver. Glutaminase breaks down glutamine into glutamate and ammonia. Glutamate also yields additional urea via the enzyme glutamate dehydrogenase. From here, ammonia is initially incorporated into hepatocyte mitochondria and ultimately results in the formation of urea. Urea subsequently leaves the hepatocyte cytoplasm and is ultimately excreted in the urine. [4][5][6][7]
Related Testing
Blood urea nitrogen (BUN) is a commonly ordered test that measures the level of urea found in blood and can assess how well the kidneys are functioning. This range may vary slightly in different healthcare settings, but a normal BUN is generally 8 to 20 mg/dL. In patients with kidney dysfunction or a high-protein diet, one can expect the BUN to be elevated. In patients with liver disease or urea cycle deficiency, one can expect the BUN to be decreased. It is important to take note of a patient’s clinical picture before assuming a patient may have renal or hepatic dysfunction.
Serum ammonia can also be measured and used to assess hepatic functioning. Normal serum ammonia generally ranges from 15 to 45 m/dL. The serum ammonia level may be elevated in a patient with hepatic dysfunction, urea cycle deficiency, overgrowth of gut flora, protein catabolism, and many other causes. Again, it is important to assess the patient’s clinical picture to identify the cause of elevated serum ammonia.
Pathophysiology
There are six clinically relevant disorders. Since all six of these disorders result in increased ammonia and decreased urea in the blood, they each have the same clinical presentation.[8][9]
Hyperammonemia is toxic to the brain and leads to encephalopathy, which can manifest as cerebral edema, vomiting, blurred vision, asterixis, and seizures. Excess ammonia will also result in the increased formation of glutamine. Recall that glutamine synthetase uses the reactants NH and glutamate to yield glutamine. Glutamate is an excitatory neurotransmitter, and therefore decreased levels of glutamate will cause depressed neural activity, which manifests as lethargy or a comatose state. In addition, excess ammonia hinders the TCA cycle by causing alpha-ketoglutarate to form glutamate.
Here we will discuss the most common urea cycle disorders:
Ornithine Transcarbamylase (OTC) Deficiency
Ornithine transcarbamylase deficiency is the only X-linked recessive enzyme deficiency in the urea cycle. It is also the most common enzyme deficiency in the urea cycle. This enzyme deficiency leads to increased levels of carbamoyl phosphate, which becomes shunted into the pyrimidine synthesis pathway once carbamoyl phosphate enters the cytoplasm. Carbamoyl phosphate is converted into orotic acid. Recall that orotic acid is the precursor of all pyrimidines. As a result, this deficiency will result in increased levels of orotic acid in the blood and urine. Orotic aciduria can manifest as orange crystals in diapers.
Argininosuccinate Synthetase Deficiency
This disorder is the second most common enzyme deficiency in the urea cycle and is autosomal recessive. Due to this enzyme deficiency, patients will have elevated levels of citrulline.
Carbamoyl Phosphate Synthetase I (CPS I) Deficiency
This disorder is also autosomal recessive but does not result in elevated orotic acid. CPS I deficiency is often fatal in infancy.
When a urea cycle enzyme deficiency is suspected, the key to identifying the source of the problem is based on the accumulation of metabolites. For example, OTC deficiency and CPS I deficiency both result in decrease urea and elevated ammonia; however, OTC deficiency will have elevated orotic acid levels, whereas CPS I deficiency will not. Another example, OTC deficiency, and hereditary orotic aciduria, both have elevated serum and urine orotic acid. One difference between these two conditions outside of their respective enzyme deficiencies is that OTC deficiency will have elevated ammonia levels, whereas hereditary orotic aciduria will not.
Clinical Significance
By understanding the biochemical pathways of the urea cycle, healthcare professionals can progress their knowledge and clinical approach in recognizing the source of elevated ammonia and decreased urea in patients with a possible urea cycle enzyme deficiency. With early detection of a urea cycle deficiency, patients, which are often newborns and infants, can be diverted from possible neurologic sequelae. In patients with a presumed urea cycle deficiency, ornithine transcarbamylase deficiency and argininosuccinate synthetase deficiency should be at the top of the differential diagnoses.