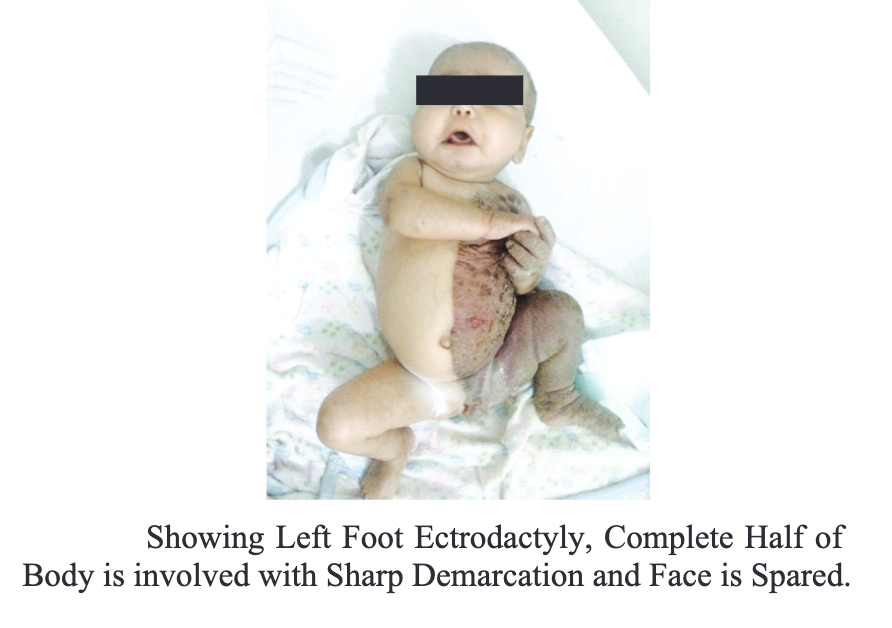
[1]
Zhuang J, Luo Q, Xie M, Chen Y, Jiang Y, Zeng S, Wang Y, Xie Y, Chen C. Etiological identification of recurrent male fatality due to a novel NSDHL gene mutation using trio whole-exome sequencing: A rare case report and literature review. Molecular genetics & genomic medicine. 2023 Mar:11(3):e2121. doi: 10.1002/mgg3.2121. Epub 2022 Dec 11
[PubMed PMID: 36504312]
Level 3 (low-level) evidence
[2]
Tan EC, Chia SY, Rafi'ee K, Lee SX, Kwek ABE, Tan SH, Ng VWL, Wei H, Koo S, Koh AL, Koh MJ. A novel NSDHL variant in CHILD syndrome with gastrointestinal manifestations and localized skin involvement. Molecular genetics & genomic medicine. 2022 Jan:10(1):e1848. doi: 10.1002/mgg3.1848. Epub 2021 Dec 26
[PubMed PMID: 34957706]
[3]
Tang MM, Tan WC, Surana U, Leong KF, Pramano ZAD. CHILD syndrome in a Malaysian adult with identification of a novel heterozygous missense mutation NSDHL c.602A}G. International journal of dermatology. 2021 Apr:60(4):e154-e156. doi: 10.1111/ijd.15296. Epub 2020 Nov 10
[PubMed PMID: 33169834]
[4]
Bittar M, Happle R. CHILD syndrome avant la lettre. Journal of the American Academy of Dermatology. 2004 Feb:50(2 Suppl):S34-7
[PubMed PMID: 14726863]
[5]
Happle R, Koch H, Lenz W. The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. European journal of pediatrics. 1980 Jun:134(1):27-33
[PubMed PMID: 7408908]
[6]
Hettiarachchi D, Panchal H, Lai PS, Dissanayake VHW. Novel variant in NSDHL gene associated with CHILD syndrome and syndactyly- a case report. BMC medical genetics. 2020 Aug 20:21(1):164. doi: 10.1186/s12881-020-01094-y. Epub 2020 Aug 20
[PubMed PMID: 32819291]
Level 3 (low-level) evidence
[7]
Fink-Puches R, Soyer HP, Pierer G, Kerl H, Happle R. Systematized inflammatory epidermal nevus with symmetrical involvement: an unusual case of CHILD syndrome? Journal of the American Academy of Dermatology. 1997 May:36(5 Pt 2):823-6
[PubMed PMID: 9146558]
Level 3 (low-level) evidence
[8]
Estapé A, Josifova D, Rampling D, Glover M, Kinsler VA. Congenital hemidysplasia with ichthyosiform naevus and limb defects (CHILD) syndrome without hemidysplasia. The British journal of dermatology. 2015 Jul:173(1):304-7. doi: 10.1111/bjd.13636. Epub 2015 May 28
[PubMed PMID: 25533639]
[9]
Mi XB, Luo MX, Guo LL, Zhang TD, Qiu XW. CHILD Syndrome: Case Report of a Chinese Patient and Literature Review of the NAD[P]H Steroid Dehydrogenase-Like Protein Gene Mutation. Pediatric dermatology. 2015 Nov-Dec:32(6):e277-82. doi: 10.1111/pde.12701. Epub 2015 Oct 13
[PubMed PMID: 26459993]
Level 2 (mid-level) evidence
[10]
Yang Z, Hartmann B, Xu Z, Ma L, Happle R, Schlipf N, Zhang LX, Xu ZG, Wang ZY, Fischer J. Large deletions in the NSDHL gene in two patients with CHILD syndrome. Acta dermato-venereologica. 2015 Nov:95(8):1007-8. doi: 10.2340/00015555-2143. Epub
[PubMed PMID: 26014843]
[11]
Johnson RL, Scott MP. New players and puzzles in the Hedgehog signaling pathway. Current opinion in genetics & development. 1998 Aug:8(4):450-6
[PubMed PMID: 9729722]
Level 3 (low-level) evidence
[12]
Has C, Bruckner-Tuderman L, Müller D, Floeth M, Folkers E, Donnai D, Traupe H. The Conradi-Hünermann-Happle syndrome (CDPX2) and emopamil binding protein: novel mutations, and somatic and gonadal mosaicism. Human molecular genetics. 2000 Aug 12:9(13):1951-5
[PubMed PMID: 10942423]
[13]
Fackler N, Zachary C, Kim DJ, Smith J, Sarpa HG. Not lost to follow-up: A rare case of CHILD syndrome in a boy reappears. JAAD case reports. 2018 Nov:4(10):1010-1013. doi: 10.1016/j.jdcr.2018.10.001. Epub 2018 Nov 9
[PubMed PMID: 30456274]
Level 3 (low-level) evidence
[14]
Porter FD. Human malformation syndromes due to inborn errors of cholesterol synthesis. Current opinion in pediatrics. 2003 Dec:15(6):607-13
[PubMed PMID: 14631207]
Level 3 (low-level) evidence
[15]
Bornholdt D, König A, Happle R, Leveleki L, Bittar M, Danarti R, Vahlquist A, Tilgen W, Reinhold U, Poiares Baptista A, Grosshans E, Vabres P, Niiyama S, Sasaoka K, Tanaka T, Meiss AL, Treadwell PA, Lambert D, Camacho F, Grzeschik KH. Mutational spectrum of NSDHL in CHILD syndrome. Journal of medical genetics. 2005 Feb:42(2):e17
[PubMed PMID: 15689440]
[16]
Preiksaitiene E, Caro A, Benušienė E, Oltra S, Orellana C, Morkūnienė A, Roselló MP, Kasnauskiene J, Monfort S, Kučinskas V, Mayo S, Martinez F. A novel missense mutation in the NSDHL gene identified in a Lithuanian family by targeted next-generation sequencing causes CK syndrome. American journal of medical genetics. Part A. 2015 Jun:167(6):1342-8. doi: 10.1002/ajmg.a.36999. Epub 2015 Apr 21
[PubMed PMID: 25900314]
[17]
Hashimoto K, Prada S, Lopez AP, Hoyos JG, Escobar M. CHILD syndrome with linear eruptions, hypopigmented bands, and verruciform xanthoma. Pediatric dermatology. 1998 Sep-Oct:15(5):360-6
[PubMed PMID: 9796585]
[18]
Gregersen PA, McKay V, Walsh M, Brown E, McGillivray G, Savarirayan R. A new case of Greenberg dysplasia and literature review suggest that Greenberg dysplasia, dappled diaphyseal dysplasia, and Astley-Kendall dysplasia are allelic disorders. Molecular genetics & genomic medicine. 2020 Jun:8(6):e1173. doi: 10.1002/mgg3.1173. Epub 2020 Apr 18
[PubMed PMID: 32304187]
Level 3 (low-level) evidence
[19]
Kim CA, Konig A, Bertola DR, Albano LM, Gattás GJ, Bornholdt D, Leveleki L, Happle R, Grzeschik KH. CHILD syndrome caused by a deletion of exons 6-8 of the NSDHL gene. Dermatology (Basel, Switzerland). 2005:211(2):155-8
[PubMed PMID: 16088165]
[20]
Zeng R, Yang X, Lin L, Jiang Y. Bilateral verruciform lesions: A new CHILD syndrome presentation. Indian journal of dermatology, venereology and leprology. 2023 May-Jun:89(3):497. doi: 10.25259/IJDVL_1293_20. Epub
[PubMed PMID: 36688896]
[21]
Armina S, Heiko T, Rudolf H, Judith F, Dimitra K, Peter S. Verrucous hyperkeratosis with predominant involvement of the left side of the body and concomitant onychodystrophy in a 17-year-old girl. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2020 Sep:18(9):1054-1057. doi: 10.1111/ddg.14141. Epub 2020 Jun 8
[PubMed PMID: 32515052]
[22]
Happle R. Ptychotropism as a cutaneous feature of the CHILD syndrome. Journal of the American Academy of Dermatology. 1990 Oct:23(4 Pt 1):763-6
[PubMed PMID: 2229513]
[23]
Sonderman KA, Wolf LL, Madenci AL, Beres AL. Insurance status and pediatric mortality in nonaccidental trauma. The Journal of surgical research. 2018 Nov:231():126-132. doi: 10.1016/j.jss.2018.05.033. Epub 2018 Jun 17
[PubMed PMID: 30278919]
[24]
Rossi M, Hall CM, Bouvier R, Collardeau-Frachon S, Le Breton F, Bucourt M, Cordier MP, Vianey-Saban C, Parenti G, Andria G, Le Merrer M, Edery P, Offiah AC. Radiographic features of the skeleton in disorders of post-squalene cholesterol biosynthesis. Pediatric radiology. 2015 Jul:45(7):965-76. doi: 10.1007/s00247-014-3257-9. Epub 2015 Feb 3
[PubMed PMID: 25646736]
[25]
Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. Journal of lipid research. 2011 Jan:52(1):6-34. doi: 10.1194/jlr.R009548. Epub 2010 Oct 7
[PubMed PMID: 20929975]
[26]
Bergqvist C, Abdallah B, Hasbani DJ, Abbas O, Kibbi AG, Hamie L, Kurban M, Rubeiz N. CHILD syndrome: A modified pathogenesis-targeted therapeutic approach. American journal of medical genetics. Part A. 2018 Mar:176(3):733-738. doi: 10.1002/ajmg.a.38619. Epub 2018 Feb 2
[PubMed PMID: 29392821]
[27]
Maceda EBG, Kratz LE, Ramos VME, Abacan MAR. Novel NSDHL gene variant for congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome. BMJ case reports. 2020 Nov 2:13(11):. doi: 10.1136/bcr-2020-236859. Epub 2020 Nov 2
[PubMed PMID: 33139364]
Level 3 (low-level) evidence
[28]
Liu T, Qian G, Wang XX, Zhang YG. CHILD syndrome: effective treatment of ichthyosiform naevus with oral and topical ketoconazole. Acta dermato-venereologica. 2015 Jan:95(1):91-2. doi: 10.2340/00015555-1859. Epub
[PubMed PMID: 24696032]
Level 2 (mid-level) evidence
[29]
Kallis P, Bisbee E, Garganta C, Schoch JJ. Rapid improvement of skin lesions in CHILD syndrome with topical 5% simvastatin ointment. Pediatric dermatology. 2022 Jan:39(1):151-152. doi: 10.1111/pde.14865. Epub 2021 Nov 17
[PubMed PMID: 34787337]
[30]
Yu X, Chen L, Yang Z, Gu Y, Zheng W, Wu Z, Li M, Yao Z. An excellent response to topical therapy of four congenital hemidysplasia with ichthyosiform erythroderma and limb defects syndrome patients with an increased concentration of simvastatin ointment. Journal of the European Academy of Dermatology and Venereology : JEADV. 2020 Jan:34(1):e8-e11. doi: 10.1111/jdv.15838. Epub 2019 Oct 1
[PubMed PMID: 31374135]
[31]
Sandoval KR, Machado MCR, Oliveira ZNP, Nico MMS. CHILD syndrome: successful treatment of skin lesions with topical lovastatin and cholesterol lotion. Anais brasileiros de dermatologia. 2019 Jul 26:94(3):341-343. doi: 10.1590/abd1806-4841.20198789. Epub 2019 Jul 26
[PubMed PMID: 31365666]