Continuing Education Activity
Wilson disease (hepatolenticular degeneration) is a rare, autosomal recessive disorder caused by abnormal copper accumulation in the body particularly involving the brain, liver, and cornea. It affects 1 in 30,000 individuals and may present as weakness, abdominal pain, jaundice, personality change, seizures, etc. This activity reviews the evaluation and management of Wilson disease and explains the role of the interprofessional team in improving care for patients with this condition.
Objectives:
Review the etiology of Wilson disease.
Summarize the faulty copper excretory mechanism in the pathophysiology of Wilson disease.
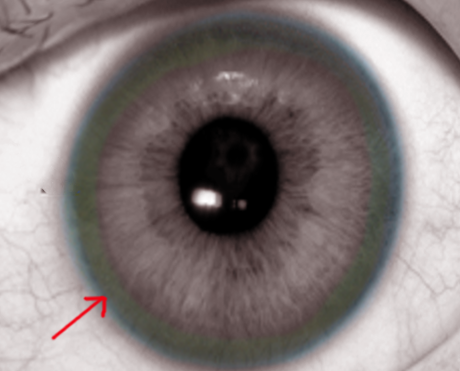
Identify the Kayser-Fleischer rings in the physical examination of patients with Wilson disease.
Outline the importance of improving care coordination among the interprofessional team to provide genetic counseling in order to prevent the transmission of the defective gene in Wilson disease.
Introduction
Wilson disease or hepatolenticular degeneration is an autosomal recessive disease which results in an excess copper build up in the body. It primarily affects the liver and basal ganglia of the brain, but it can affect other organ systems too.[1][2][3]
Symptoms usually are related to the brain and liver. Liver-related symptoms include vomiting, weakness, ascites, swelling of the legs, yellowish skin, and itchiness. Brain or neurological symptoms include tremors, muscle stiffness, trouble speaking, personality changes, anxiety, and auditory or visual hallucinations.
Wilson disease is an autosomal recessive condition caused by a mutation in the Wilson disease protein (ATP7B) gene. For a person to be affected, a copy of the gene from each parent need to be inherited. Diagnosis is difficult and involves blood tests, urine tests, and a liver biopsy along with the clinical evaluation. Genetic testing may be used to screen the family members of those affected.
The genetic defect is localized to long arm of chromosome 13 (13q), which has been shown to alter the copper transporting ATP gene in the liver. The majority of patients with Wilson disease present within the first decade of life with liver dysfunction. The neuropsychiatric features are seen in the third/fourth decade of life. Wilson disease is rare but if not recognized and treated, it is fatal.
Etiology
Wilson disease is caused by one of the several mutations in ATP7B gene present on chromosome 13 which controls the protein transporter responsible for excreting excess copper into bile and out of the body. The protein transporter is located in the trans-Golgi network of the liver and brain. The major route of copper excretion (95%) is through the liver. This excess copper first accumulates in the liver and then spills into the blood and then to other organ systems.[4]
The excess copper leads to the generation of free radicals that causes oxidation of vital proteins and lipids. Early changes tend to occur in the mitochondria, nuclei, and peroxisomes.
Epidemiology
This disease affects 1 in every 30,000 individuals with a carrier frequency of 1 in every 90. Some populations have a higher incidence of Wilson disease due to the increased rate of consanguinous marriages. Both males and females are affected equally. The usual age of presentation is four to 40 years of age, but this disorder has been detected in children as young as three as well as adults as old as 70.
Pathophysiology
In Wilson disease, there is a faulty copper excretory mechanism, causing copper to accumulate in the liver and spill into the blood where it begins to accumulate in other organs and tissues like subthalamus, putamen, and cortex of the brain, kidneys, and cornea. Copper is a transition metal, and excess copper levels lead to the formation of a toxic hydroxyl group and increase oxidative stress in the cells. This oxidative stress damages the cells and leads to the clinical manifestation, namely liver failure, behavioral problems, movement disorders, and Kayser-Fleischer rings in the cornea.[5]
Copper is needed by the body, predominantly as a cofactor for some enzymes such as ceruloplasmin, cytochrome c oxidase, dopamine beta-hydroxylase, superoxide dismutase, and tyrosinase. Copper enters the body through the digestive tract with the help of a transporter protein in the cells of the small bowel, copper membrane transporter 1 (Ctr1; SLC31A1). This transporter helps to carry copper inside the cells where part of the copper is bound to metallothionein and part is carried by ATOX1 to an organelle known as the trans-Golgi network. In response to rising copper levels, an enzyme called ATP7A releases copper into the portal vein to the liver. Liver cells carry the CMT1 protein and metallothionein, and then ATOX1 binds it inside the cell. Once here, ATP7B links the copper to ceruloplasmin and releases it into the bloodstream, removing the excess copper by secreting it into bile. Both functions of ATP7B are dysfunctional in Wilson disease. Copper accumulates in the liver, and ceruloplasmin is secreted in a form that lacks copper and is rapidly degraded in the bloodstream.
When the copper level in the liver overwhelms the proteins that normally bind it, it results in oxidative damage through a process known as Fenton chemistry. This damage results in chronic active hepatitis, fibrosis, and cirrhosis. The liver releases copper into the bloodstream that is not bound to ceruloplasmin. This free copper precipitates throughout the body, particularly in the kidneys, eyes, and brain. In the brain, copper is deposited in the basal ganglia, putamen, and globus pallidus (i.e., lenticular nucleus); these areas participate in the coordination of movement and neurocognitive processes such as stimulating mood regulation. Damage to these areas produces the neuropsychiatric symptoms seen in Wilson disease.[6]
Histopathology
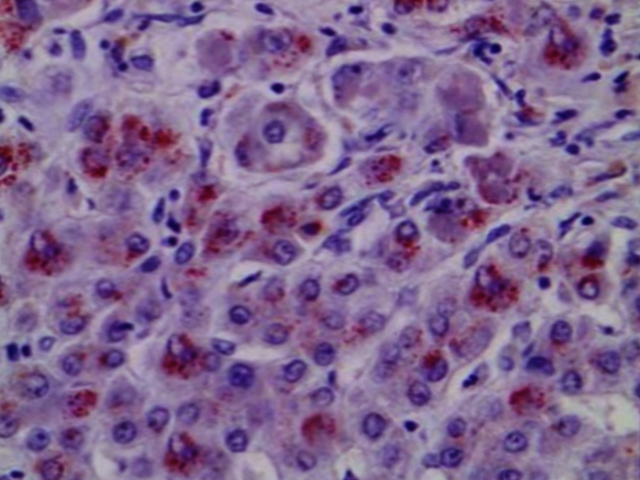
Light microscopy may show early changes including moderate fatty infiltration and glycogen deposition in the hepatocytes. The histological features may resemble chronic active hepatitis. Even though copper levels are elevated in the liver, early on the copper is located in the cytoplasm and may not be apparent with rhodamine staining.
History and Physical
Since Wilson disease is a heritable disorder, patients may have a positive family history. Patients may complain of abdominal pain, jaundice, weakness, personality changes, depression, migraine headaches, insomnia, seizure, and movement disorder chorea. Hemiballismus may also be present in history.
About 30-50% of patients will have neuropsychiatric symptoms including asymmetrical tremor. Other symptoms may include drooling, ataxia, personality changes, mask-like facies, and clumsiness.
On physical exam, the patient may have features of hepatosplenomegaly, isolated splenomegaly, or if the disease has progressed to cirrhosis, then stigmata of chronic liver disease may also be prominent. An eye exam may reveal icterus of sclerae and slit-lamp examination for Kayser-Fleischer (KF) rings on the cornea (note that the only other disorder with KF rings is Primary biliary cirrhosis). Other features of Wilson disease may include the presence of movement disorders, difficulty speaking, mask-like faces, spasticity, muscle rigidity, and skin findings of lunulae ceruleae (bluish discoloration at the base of fingernails).
Skeletal involvement is very common in Wilson disease and resembles premature osteoarthritis. The arthropathy usually involves the axial skeleton and spine.
Hemolytic anemia is seen in 10-15% of patients and is due to red cell lysis by the high copper concentration. When clinicians are faced with a young patient with impaired liver function, hemolytic anemia, and normal alkaline phosphatase levels, Wilson disease should be suspected.
Renal symptoms are similar to Fanconi syndrome and urolithiasis.
Evaluation
If suspicion of Wilson disease is high, order a ceruloplasmin level. It will be less than 20 mg/dL (normal 20 mg/dL to 40 mg/dL). Urinary copper levels will be raised more than 100 mcg/dL. These two lab findings with Kayser-Fleischer rings are usually enough for diagnosis, but if there is the possibility of an alternate diagnosis, order a liver biopsy for liver copper levels; this the most accurate test for Wilson disease. Note low levels of ceruloplasmin may be seen in any protein deficiency disorder.
A positive result is a copper level greater than 250 mcg/g of dry liver tissue. An MRI is helpful to check for brain involvement. Liver function tests are deranged with elevated AST and ALT levels.[2][7]
Wilson disease should be suspected if symptoms consistent with the disease are present or if a relative has been found to have the disease. Most had slightly abnormal liver function tests and raised aspartate transaminase, alanine transaminase, and bilirubin levels. If the liver damage is significant, albumin is decreased as a result of the inability of damaged liver cells to produce this protein; likewise, the prothrombin time is prolonged as the liver is not producing proteins known as clotting factors. Alkaline phosphatase levels are low in those with Wilson-related acute liver failure. If there are neurological symptoms, MRI of the brain may show hyperintensities in the basal ganglia in the T2 sequence. MRI may demonstrate the characteristic "face of the giant panda" pattern.
The ECG may reveal ventricular hypertrophy, arrhythmias and non-specific changes in T waves and ST segments.
The presence of Kayser Fleischer rings in the presence of neuropsychiatric symptoms is suggestive of Wilson disease.
There is no completely reliable test for Wilson disease, but levels of ceruloplasmin and copper in the blood, as well as copper excreted in urine during a 24-hour period, are used to form an impression of the amount of copper in the body. The gold standard is a liver biopsy.
First and second-degree relatives need to be screened for Wilson disease.
Treatment / Management
The mainstay therapy for Wilson disease is copper chelation therapy with penicillamine and trientine. Trientine is preferred because of fewer side effects. Oral zinc also may be given as it competes for absorption with copper at metallic ion transporter. It is vital to educate the patient on the adverse effects of chronic chelation therapy which can make symptoms worse. D-penicillamine can be used during pregnancy and does not pose any risk to the fetus
If the patient develops liver cirrhosis and its associated complication, Transjugular intrahepatic portosystemic shunt(TIPS) can be offered for recurrent variceal bleed. Liver transplantation is curative.[8][9][10]
For muscle rigidity, spasticity, and parkinsonian features, baclofen, anticholinergics(trihexyphenidyl), GABA antagonists, and levodopa may be used.
Liver transplantation seemed to have been beneficial in improving neurological dysfunction in some patients not responding adequately to medical therapy in a French study[11]
A diet low in copper-containing foods is recommended with avoidance of mushrooms, chocolate, nuts, dried fruit, liver, and shellfish.
Physiotherapy and occupational therapy are beneficial in the neurologic form of the disease. The copper-chelating treatment takes up to six months to start working, and these therapies can assist in coping with ataxia, dystonia, and tremors as well as preventing contractures that can result from dystonia.
Differential Diagnosis
Differential diagnosis includes conditions causing chronic active hepatitis, hemolytic anemias due to various causes.
Neuropsychiatric disorders that can be mistaken include various types of Parkinsonian syndromes, pantothenate kinase deficiency associated neurodegeneration(iron accumulation), neuroacanthocytosis syndromes and Huntington's disease to name a few. These conditions may present with personality changes, rigidity, dystonias and movement disorders.
Staging
Staging of Wilson Disease
- Stage 1: initial accumulation of copper in the liver
- Stage 2: Acute redistribution of copper within the liver, followed by release into the systemic circulation
- Stage 3: Chronic accumulation of copper into extrahepatic tissues including brain
- Stage 4: Use of chelation therapy to restore copper balance
Prognosis
Various scoring systems have been put forward for prognostication. Some of these incorporate levels of AST, bilirubin, and prothrombin time, etc. on presentation. [12]
Patients with a prognostic score of 7 or more should be referred for a liver transplant. Patients with this score are generally dead within 8 weeks without treatment. After liver transplant, the prognosis is good. A survival rate of 87% at 15 years has been reported [13]
Complications
- Liver failure
- Encephalopathy
- Splenomegaly
- Ascites
- Variceal bleeding
- Hepatorenal syndrome
- Liver failure
- Neuropsychiatric manifestations
- Death
Postoperative and Rehabilitation Care
Long term monitoring is vital with measurements of urinary copper, complete blood counts, free copper, renal and liver function at 4-8 week intervals. The eyes should also be examined. Most patients need lifelong chelation therapy with follow up. Patients need to avoid alcohol and all hepatotoxic medications.
Consultations
Services of ophthalmology, gastroenterology, hematology, neurology, and psychiatry, as well as social services, should be consulted for optimizing care of these patients
Pearls and Other Issues
Wilson disease, if untreated, will eventually lead to chronic liver disease and may even progress to hepatocellular carcinoma in some cases. Genetic counseling is necessary to prevent the transmission of the defective gene.
Enhancing Healthcare Team Outcomes
Wilson disease has no specific curative treatment and is progressive. Without treatment it is fatal. The disorder is best managed by an interprofessional team that includes a gastroenterologist, geneticist, mental health nurse, neurologist, dietitian, nurse practitioner, pathologist, radiologist, and an internist.
The mainstay therapy for Wilson disease is copper chelation therapy with penicillamine and trientine. Some patients with early disease may be offered liver transplantation. However, it is the neurological features of the disease that are difficult to manage. The patient should be educated on the consumption of low copper-containing foods. The pharmacist should ensure that the patient is on no medication that can adversely affect the liver. Consumption of alcohol should be forbidden. The mental health nurse should periodically assess the neuropsychiatric symptoms, which may require additional drug treatments. While liver transplantation is curative, patients need to be monitored for the adverse effects of the immunosuppressant drugs. The pharmacist needs to monitor the chelation therapy because it also has many adverse effects that can worsen the symptoms. Sometimes, chelation therapy has to be discontinued. A team approach involving nursing, pharmacists, and clinicians will improve outcomes. [Level 5]
Physiotherapy and occupational therapy are beneficial in the neurologic form of the disease. Genetic counseling by a genetics nurse and clinician geneticist is necessary to prevent the transmission of the defective gene. Only with such a team approach can the mortality and morbidity of Wilson disease be reduced.
The outlook for most patients is guarded because there is no cure. The quality of life of most patients is poor. [14][15][16](Level 5)