Continuing Education Activity
Neurofibromatosis is a neurocutaneous disorder characterized by tumors in the nervous system and skin. The most common types of neurofibromatosis are types 1 and 2, both of which are autosomal dominant. Neurofibromatosis type 1, also known as von Recklinghausen disease, presents with neurofibromas, cafe-au-lait spots, freckling, and optic gliomas. Neurofibromatosis type 2 is characterized by bilateral vestibular schwannomas and meningiomas. Treatment for neurofibromatosis types 1 and 2 involves clinical monitoring and medical intervention when appropriate. This activity describes the pathophysiology, evaluation, and management of neurofibromatosis and highlights the role of the interprofessional team in caring for patients affected by this condition.
Objectives:
Identify the etiology of neurofibromatosis.
Describe the presentation of a patient with neurofibromatosis.
Summarize the treatment and management options available for neurofibromatosis.
Explain interprofessional team strategies for improving care coordination and communication to advance the treatment of neurofibromatosis and improve outcomes.
Introduction
Neurofibromatosis is a neurocutaneous disorder characterized by tumors in the nervous system and skin. Neurofibromatosis types 1 and 2 are the most common and are distinct entities. Neurofibromatosis type 1, or von Recklinghausen disease, is an autosomal dominant disease. Neurofibromatosis type 1 presents with neurofibromas, cafe-au-lait macules, freckling, and optic gliomas. It is a clinical diagnosis. Neurofibromatosis type 2 (NF2) is a disease characterized by bilateral vestibular schwannomas (VS) and meningiomas. It has an autosomal dominant inheritance pattern. Treatment for neurofibromatosis types 1 and 2 is clinical monitoring and medical intervention when appropriate.[1][2][3]
Etiology
Neurofibromatosis type 1 is caused by a loss of function mutation, either de novo or inherited, on the neurofibromin 1 (NF1) gene. It is located on band 17q11.2 and codes for neurofibromin. Neurofibromin is a tumor suppressor that functions in the RAS/MAPK and mTOR pathways. Mosaicism can occur, resulting in the segmental, generalized, or gonadal NF1 gene. Segmental NF1 gene has pigment changes, tumors, or both that are limited to one or more body segments. Generalized NF1 gene appears similar to classic NF1 but does not have the NF1 gene mutation. Gonadal NF1 gene occurs when the mutation only affects the ova or sperm.
NF2 gene is caused by a loss of function mutation of the NF2 gene. It is located on band 22q12 and codes for merlin. Merlin is a cell membrane protein that is also a tumor suppressor that functions in the PI3kinase/Akt, Raf/MEK/ERK, and mTOR pathways. [4][5]
Epidemiology
Neurofibromatosis type 1 makes up about 96% of all neurofibromatosis cases. Prevalence is 1 in 3000 births. It occurs equally between gender and races. Fifty percent of patients have a spontaneous mutation, and the other half have an inherited mutation. There is a 100% penetrance with variable expressivity. Neurofibromatosis type 2 makes up about 3% of all cases and has a prevalence between 1 in 33,000 births and 1 in 87,410. There is no gender or race predilection. Neurofibromatosis type 2 has variable presentations amongst different families. A more severe clinical presentation is associated with a frameshift or a nonsense mutation that results in a truncated protein. [6]
Histopathology
Neurofibromas are benign tumors with mixed cell types, including Schwann cells, perineural cells, and fibroblasts. The tumors also contain mast cells, axonal processes, and a collagenous extracellular matrix.
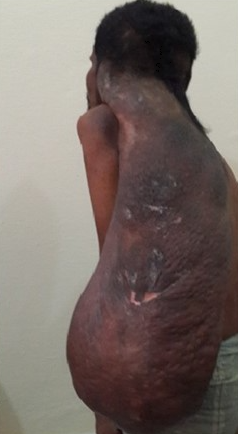
Plexiform neurofibromas are similar to neurofibromas, but they arise from muscle nerve fascicles and can infiltrate into the surrounding structures.
Schwannomas arise from the eighth cranial nerve. They are composed of spindle cells with mixed Antoni A and Antoni B cellular arrangements, verocay bodies, and hyalinised vessels. [7][8]
History and Physical
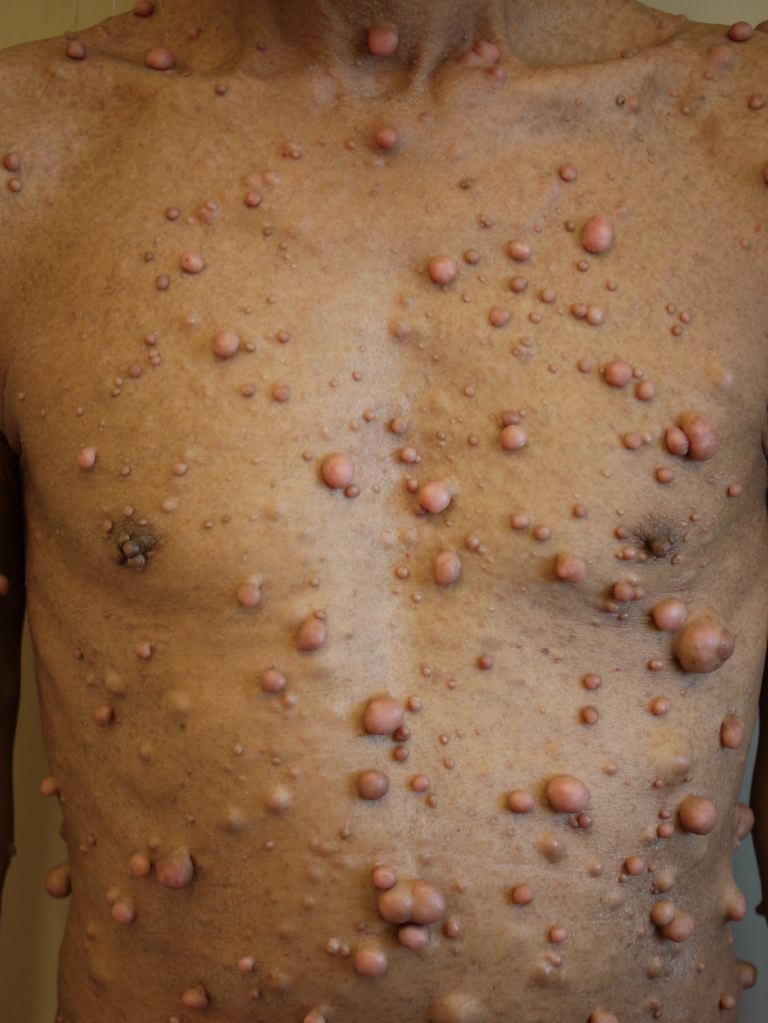
Neurofibromatosis type 1 has cutaneous and noncutaneous manifestations. Cafe-au-lait macules are one of the seven diagnostic criteria for neurofibromatosis type 1. The lesions are sharply demarcated with a homogenous appearance. Axillary and groin freckling, or Crowe sign, is the most specific criteria for neurofibromatosis type 1. Neurofibromas can occur anywhere on the body. They can be cutaneous or internal. Dermal tumors are soft dome-shaped tumors but can also present as pedunculated, nodular, or plaque-like. Internal tumors are deeper and can occur around the eye, retroperitoneal, along with the gastrointestinal (GI) tract, or in the mediastinum. Neurofibromas have the buttonhole sign. Plexiform neurofibromas are usually present from birth and are derived from the nerve sheaths. They can feel like a "bag of worms." Cutaneous manifestation includes scoliosis, long bone dysplasia, learning difficulties, and attention deficit hyperactivity disorder (ADHD). Lisch nodules are hyperpigmentation in the iris. They do not affect vision. Optic glioma is a tumor of the optic nerve and can affect vision. It occurs in 15% of patients with neurofibromatosis type 1. Patients also have generalized hyperpigmentation, blue-red, pseudoatrophic macules, juvenile xanthogranuloma, glomus tumor, melanoma, nevus anemicus, and pruritus. Patients are at increased risk for rhabdomyosarcoma, myeloid leukemia, and pheochromocytomas.
Neurofibromatosis type 2 patients present most commonly with schwannomas and meningiomas. Bilateral vestibular schwannoma and unilateral vestibular schwannoma occur on the superior division of the eighth cranial nerve. This is the most common type, but it can occur with any cranial nerve. Involvement of the facial nerve with the vestibular schwannoma can make surgical treatment difficult. These patients present with tinnitus, hearing loss, and difficulty with balance. Patients that have the truncated protein were found to have the disease of onset at a younger age and a higher prevalence of tumors. Younger patients also have an earlier onset of symptoms.[9]
Evaluation
NIH has seven diagnostic criteria for neurofibromatosis type 1. Two must be met for the diagnosis of neurofibromatosis type 1. Genetic testing is not routinely done.
- Six or more cafe-au-lait spots greater than 5 mm prepubertal and greater than 15 mm post-pubertal
- Two or more neurofibromas or one or more plexiform neurofibroma
- Axillary or groin freckling
- Optic glioma
- Two or more Lisch nodules
- Sphenoid dysplasia, dysplasia or thinning of long bone cortex
- First-degree relative with neurofibromatosis type 1
Differential diagnosis includes neurofibromatosis type 1-like syndrome, familial cafe-au-lait spots, and segmental neurofibromatosis type 1. The nf1-like syndrome was first described in 2007. Patients with the neurofibromatosis type 1-like syndrome have cafe-au-lait spots, axillary freckling, and macrocephaly, but they lack the NF1 genetic mutation, neurofibromas, and Lisch nodules. It is an autosomal dominant disease due to a mutation in the SPRED1 gene on chromosome 15. Familial cafe-au-lait spots is a disorder presenting with only cafe-au-lait macules.
Bilateral vestibular schwannoma is pathognomic for neurofibromatosis type 2, but not all patients with neurofibromatosis type 2 have bilateral vestibular schwannoma. The NIH has diagnostic criteria for neurofibromatosis type 2.
Definitive Neurofibromatosis Type 2
- Bilateral vestibular schwannoma or a first-degree relative with neurofibromatosis type 2 plus unilateral vestibular schwannoma in someone less than 30 or any two of the following: meningioma, glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
Presumptive or Probable Neurofibromatosis Type 2
- Unilateral vestibular schwannoma in someone under 30 plus one of the following: meningioma, schwannoma, glioma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
- Multiple meningiomas (2 or greater) plus unilateral vestibular schwannoma less than 30 years or one of the following: schwannoma glioma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract [10]
Treatment / Management
Cafe-au-lait spots and neurofibromas are benign and do not require treatment. Surgical excision can be performed on symptomatic lesions, but the recurrence can occur. Plexiform neurofibromas have malignant potential. There is an 8% to 13% risk for plexiform neurofibromas to develop into malignant peripheral nerve sheath tumors. This should be suspected if there is pain for more than one month, new neurologic deficits, change of the neurofibroma from soft to hard, or a rapid increase in size. These malignancies are treated with wide local excision. Imatinib has been shown to decrease plexiform neurofibroma size.
Monitoring for any neurologic changes and referral to a neurologist is paramount. These changes can be due to tumor development. Consistent ophthalmologic evaluation is recommended for observation of the development of optic gliomas. Chemotherapy is the treatment of choice for optic gliomas.
Monitoring children for difficulty learning and behavioral issues is important. Counseling can be beneficial for patients to provide support regarding the disease's autosomal dominant inheritance pattern.
Neurofibromatosis type 2
Patients with neurofibromatosis type 2 require the assessment of their hearing. Ophthalmology evaluation, MRI, audiology, and brainstem evoked potentials are important in managing these patients. Surgery is still the first-line treatment for symptomatic tumors, but there is a 44% recurrence rate. Radiation can be used, but there is an increased risk of malignant transformation. Bevacizumab, a VEGF inhibitor, is a monoclonal antibody and can be used to treat neurofibromatosis type 2 patients medically. It decreased tumor size in 53% of cases and improved hearing in 57%. Patients with suspected neurofibromatosis type 2 should have an MRI of the head and spine done. Getting thin cuts through the internal auditory canals is important. Treatment is done if the tumor is compressing the brainstem or preventing hearing loss. [11][12][13]
Differential Diagnosis
- Acoustic neuroma
- Brainstem syndromes
- Café-au-lait spots
- Legius syndrome (SPRED1 -related café-au-lait spots and freckles)
- McCune-Albright syndrome
- Spinal injury
Enhancing Healthcare Team Outcomes
The management of neurofibromatosis is best done with an interprofessional team, including dermatologists, neurologists, pediatricians, and genetic counselors. The key is to monitor the patient for CNS tumors, which, if detected late, have a poor prognosis. Genetic counseling should be available to patients with an affected child. The dermatologist, ophthalmologist, neurosurgeon, neurologist, and general surgeon should regularly follow up with these patients to ensure that no mass lesions are developing in the body.