Continuing Education Activity
As the name implies, neurocutaneous syndromes are disorders involving the nervous system and the skin. Two of the most common neurocutaneous syndromes also referred to as phakomatoses, are neurofibromatosis (NF) and tuberous sclerosis complex (TSC). Neurofibromatosis and tuberous sclerosis complex are inherited in an autosomal dominant manner, though the expressivity of these diseases varies. This activity reviews neurocutaneous syndromes, with particular focus on neurofibromatosis (NF) and tuberous sclerosis complex (TSC). This activity highlights the role of the interprofessional team in the care of affected patients.
Objectives:
- Review features that should prompt consideration of a neurocutaneous syndrome.
- Outline the diagnostic criteria for neurofibromatosis (NF) and tuberous sclerosis complex (TSC).
- Summarize the treatment of neurofibromatosis (NF) and tuberous sclerosis complex (TSC).
- Explain the need for optimizing care coordination, with particular emphasis on communication between interprofessional medical teams, to enhance prompt evaluation and management of neurocutaneous syndromes and decrease the risk of complications and improve outcomes.
Introduction
As the name implies, neurocutaneous syndromes are disorders involving the nervous system and the skin. It is essential for clinicians, especially pediatricians, to recognize the skin findings associated with neurocutaneous syndromes because this will guide in the surveillance for and management of possible complications associated with the condition.
Two of the most common neurocutaneous syndromes also referred to as phacomatoses, are neurofibromatosis (NF) and tuberous sclerosis complex (TSC).
Etiology
In NF, tumors of the nerve sheaths develop. A mutation in the NF1 gene, on chromosome region 17q11.2, encoding neurofibromin, has been identified.[1] Neurofibromin is an inhibitor of the oncogene Ras, and impaired function of this gene will lead to overactivation of Ras, giving rise to the development of tumors. The mode of inheritance for NF is autosomal dominant, but 50% of the cases result from a de novo mutation.
The NF2 gene encodes a protein called merlin or schwannomin, located on chromosome 22q1.11.[2] The condition is also inherited in an autosomal dominant fashion, with approximately half of the cases caused by a de novo mutation.
In a patient with TSC, a mutation in either the TSC1 (9q34.13) or TSC2 (16p13.3) gene is the underlying defect.[3] This disorder also has an autosomal dominant inheritance pattern, with complete penetrance, but with variable expression. Again, at least 50% of the cases are sporadic, being the consequence of a de novo mutation.
Epidemiology
Neurofibromatosis type 1 (NF1) is the most common of all the neurocutaneous syndromes, with a prevalence of 1:3,000 compared to Neurofibromatosis type 2 (NF2) with a prevalence of 1:25,000. Tuberous Sclerosis Complex (TSC) has a prevalence of 1:6,000 individuals worldwide. There is no predilection for ethnicity, gender, race or geography in any of these disorders.[4]
Pathophysiology
The following proteins synthesized by the respecitve genes become faulty in various neurocutaneous syndromes:
- NF1 - Neurofibromin
- NF2 - Merlin
- TSC1 - Hamartin
- TSC2 - Tuberin
History and Physical
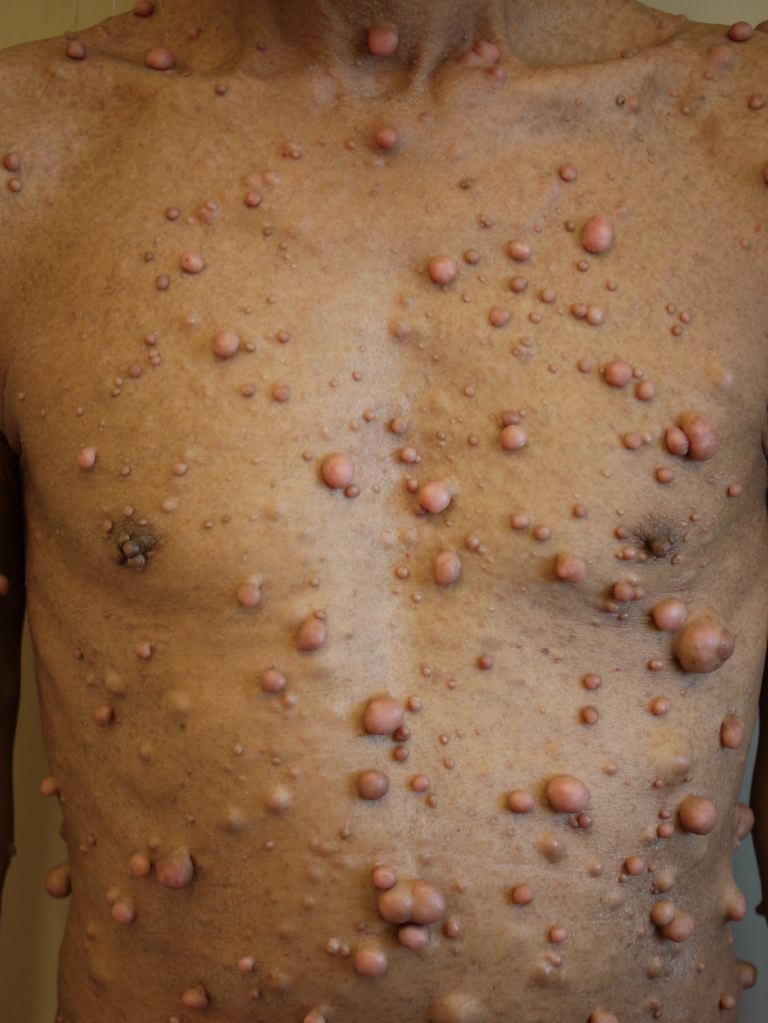
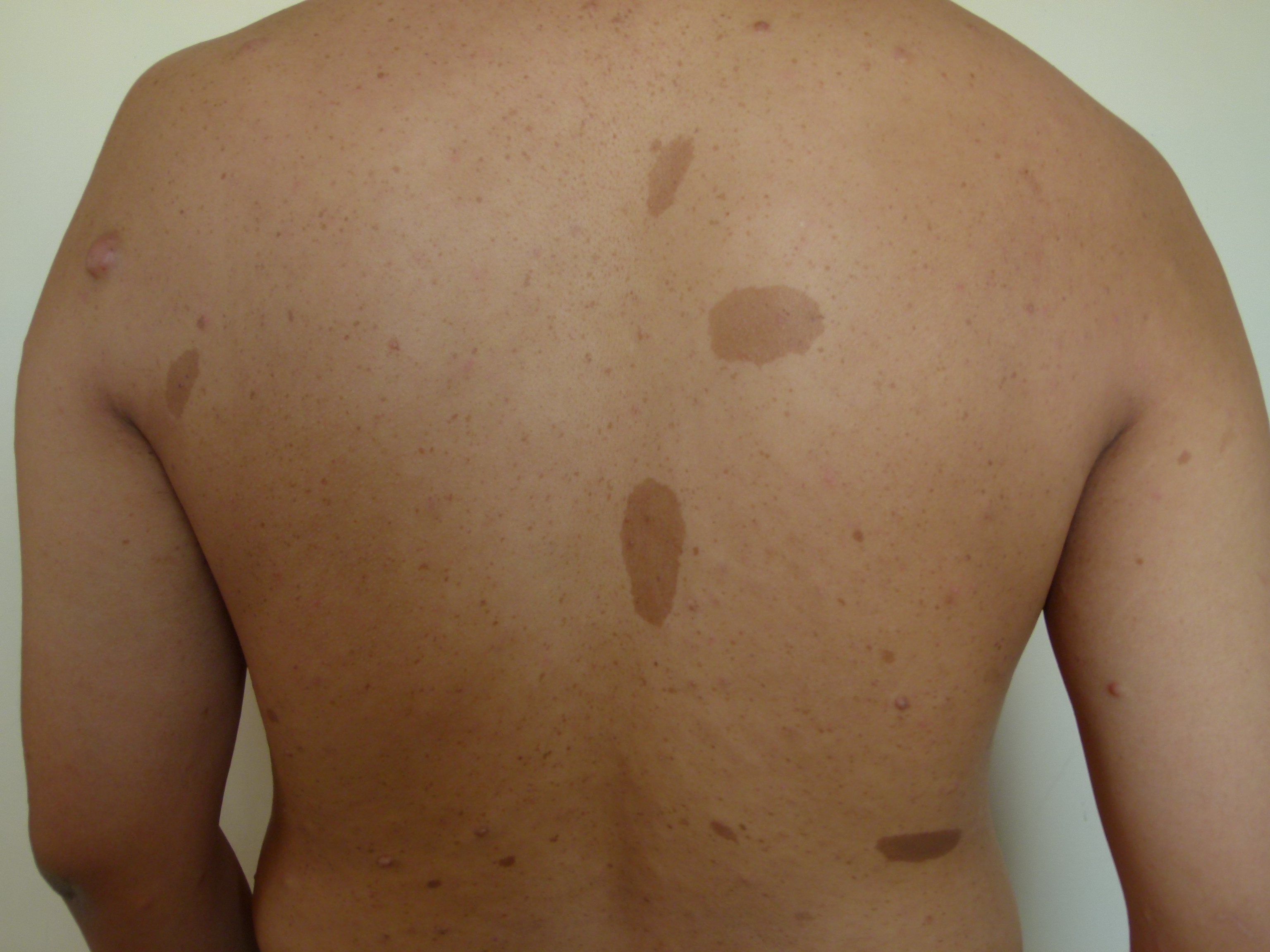
The chronology of the presenting signs of NF1 are important to keep in mind, and for the pediatrician to suspect NF1, the physical examination is critical, especially for the cutaneous manifestations. The hallmark of NF1, and usually the first presenting signs, are the café-au-lait macules. These macules are common in the general population, but if a patient has 6 or more large café-au-lait macules, the clinician should actively seek for other criteria of NF1. At around 3-5 years of age, patients may develop and present with freckling, most commonly in the axillary and inguinal area, but also in the nape of a person’s neck and around the mouth. The benign tumors that give rise to the name of NF are neurofibromas. These are benign Schwann cell tumors of the nerve sheath. Young children with NF1 can have plexiform neurofibromas, most commonly in the regions of the eye, neck, or limbs. Dermal fibromas, which are smaller and can present in larger quantities, develop during puberty and onwards.[1][4]
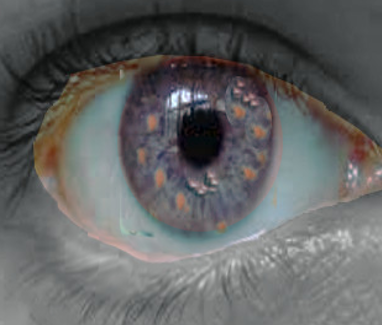
The non-cutaneous manifestations to look out for in NF1 include optic glioma, although these most commonly remain asymptomatic. If symptoms occur, they can cause loss of vision or disturbances of the hypothalamus, when the location of the tumor is in the vicinity of the optic chiasm. If an optic glioma develops, it most commonly will at around 2-6 years of age. The manifestation of NF1 in the eye itself are Lisch nodules, which are hamartomas in the iris. It is essential to realize though, that these usually present later in life, as the prevalence increases with age. Nearly all patients with NF1 will have Lisch nodules by 21 years of age. Another sign that could raise the concern of NF1 is tibial dysplasia, which is congenital, but the problem’s identification is possible only when the patient begins to stand.[1][4][5][4]
NF2 is less common than NF1, and there are not as many dermatologic manifestations. The manifestations that define NF2 are tinnitus, hearing loss, loss of balance, and sometimes facial nerve paralysis, secondary to bilateral vestibular schwannomas. Patients can have café-au-lait macules, but it’s not as much of a hallmark of the syndrome as it is for NF1. Patients with NF2 have no freckling or cutaneous neurofibromas. They can, though, develop plaque-like skin growths called schwannomas.[2][4][6][4]
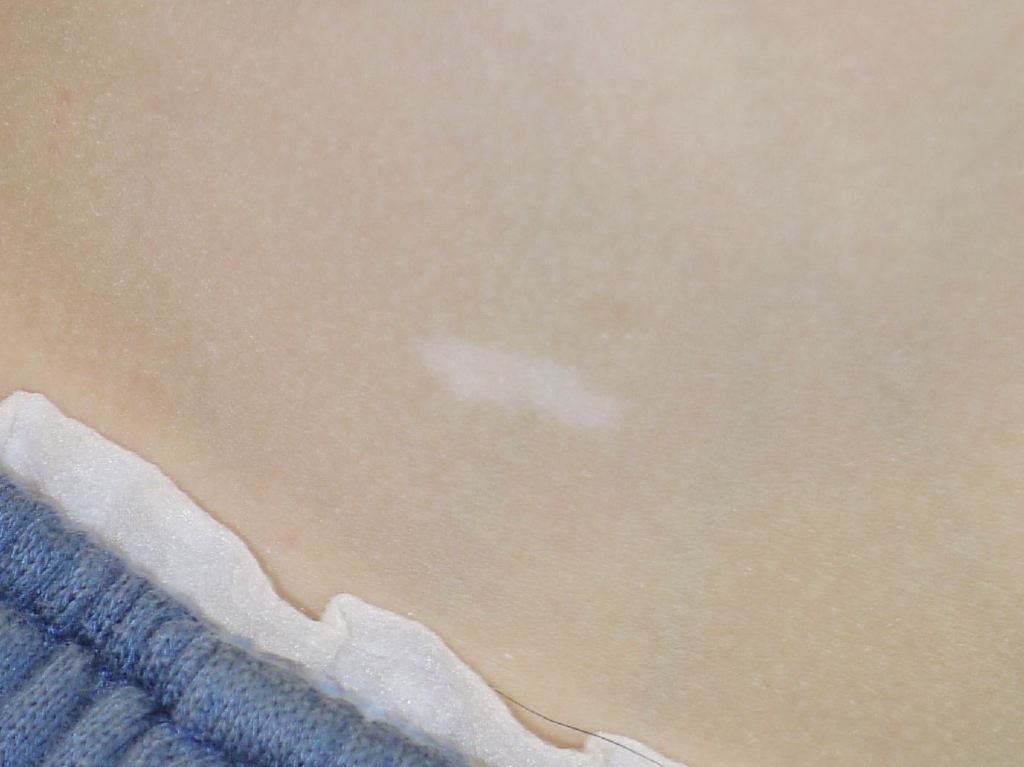
As in neurofibromatosis, TSC has specific cutaneous manifestations that should prompt the clinician to think of this disorder. Patients will present with hypopigmented macules, frequently referred to as “ash-leaf” spots because of their shape. These macules are congenital, although they may not be visible or identifiable at birth, and are identifiable with a Wood’s lamp in a dark room. If three or more hypopigmented macules are present, the suspicion for TSC is high.
Similar to NF, it is important to understand the timeline of manifestations in TSC. Between four and six years of age, small red nodules over the nose and cheeks may appear, which are facial angiofibromas. On the lower back, there may be a rough and raised plaque with an orange peel-like surface, called a shagreen patch. Around 15-20% of patients with TSC will develop ungual fibromas around adolescence.[3][4]
TSC also has non-cutaneous manifestations, including cardiac rhabdomyomas, which are congenital and are likely to regress spontaneously but may cause obstruction in the heart of a newborn. Other important manifestations are infantile spasms or seizures, which often start right before the patient’s second birthday. Early recognition and treatment are of utmost importance because its delay may lead to severe intellectual disability.[7]
Evaluation
When a clinician detects the findings described above, a thorough exam is mandatory to start the search for other signs that could help in making the diagnosis of a neurocutaneous disorder. The diagnosis of neurocutaneous syndromes is clinical, and studies are usually helpful to confirm the diagnosis and to counsel the family. To diagnose a patient with NF1, the patient has to meet 2 or more criteria. These criteria are six or more café-au-lait macules larger than 5 mm in prepubertal children or larger than 15 mm after puberty, skinfold freckling, two or more neurofibromas, or one plexiform neurofibroma, long bone or sphenoid wing dysplasia, optic glioma, Lisch nodules or a first-degree relative with NF1.[8][9]
A child with a risk for NF 2, because of a first-degree relative affected with the disorder, should undergo evaluation by an audiologist, followed up with a head magnetic resonance imaging (MRI) if any abnormalities. A child diagnosed with NF2 requires at least an MRI of the brain by adolescence.
TSC is also a disorder in which the diagnosis is clinical. For the clinician to make the diagnosis, the patient has to meet two major criteria or 1 major and 2 or more minor criteria. The major criteria are three or more hypopigmented macules of at least five mm of diameter, three or more angiofibromas or a fibrous cephalic plaque, two or more ungual fibromas, a shagreen patch, multiple retinal hamartomas, cortical dysplasias, subependymal nodules, a subependymal giant cell astrocytoma, a cardiac rhabdomyosarcoma, lymphangioleiomyosarcomatosis and two or more angiomyolipomas. Minor criteria are: several small hypopigmented lesions grouped together, also called “confetti” skin lesions, three or more dental enamel pits, two or more intraoral fibromas, a retinal achromic patch, multiple renal cysts, and nonrenal hamartomas.
Treatment / Management
Once the diagnosis of NF1 has been established, the most important component of management is surveillance for complications associated with the syndrome. For the clinician to understand this, there must be an understanding of the natural history of NF1 as described above in the manifestations. Complex neurofibromas can grow, becoming symptomatic by compressing the spinal cord or airway, prompting referral for surgical management. Because of the increased risk for optic glioma, patients require annual ophthalmologic evaluation, at least until they are 6-7 years of age since optic glioma is unlikely to develop after that age. If the patient develops symptoms from an optic glioma, the treatment commonly is chemotherapy. Monitoring blood pressure is vital because renal artery stenosis or pheochromocytoma could lead to hypertension. Once the patient stands or ambulates, around 10 to 12 months of age, long bone dysplasia, more commonly tibial dysplasia, may be more evident and radiographs will show the findings. Once confirmed, the patient should receive a prompt referral to orthopedics for further management. It is of great importance to recognize NF1 early, due to the neurocognitive dysfunction that at least half of the patients will develop, most commonly learning disabilities. Early recognition of these neurocognitive problems, leading to early intervention, will give the patient the best opportunities available to develop and function well into adulthood.
In patients with NF1, there is also an increase in the incidence of pheochromocytoma, rhabdomyosarcoma, leukemia, and Wilms tumor, compared with the general population.[10] Due to increased risk for scoliosis, patients should have a yearly screening.
In NF2, the need for treatment is only if the tumors become symptomatic, and the treatment of choice would be surgery.
For TSC, it is very important as well to identify the disorder early, especially if the patient develops infantile spasms or seizures because those warrant timely instauration of treatment. The goal is to avoid intractable seizures and to prevent severe cognitive impairment.[11] Follow up of patients with TSC is mostly about surveillance and treatment of complications. The recommendation is to do a brain MRI and kidney imaging every 1-3 years. In case of signs or symptoms of increased intracranial pressure, one must suspect an obstruction of the foramen of Monro by a subependymal giant cell astrocytoma. Brain images are to be performed immediately, with referral to neurosurgery for further management.
Differential Diagnosis
Neurocutaneous syndromes include a wide variety of diagnoses and differential diagnoses:
- Neurofibromatosis
- Schwannomatosis
- Sturge-Weber syndrome
- Tuberous sclerosis complex
- Von Hippel-Lindau disease
Prognosis
Prognosis varies with the age at presentation and the associated lesions, including malignancies.
Complications
Patients with neurofibromatosis type 1 are likely to develop tumors like malignant peripheral nerve sheath tumor, gastrointestinal stromal tumor, optic pathway glioma, pheochromocytoma, and juvenile myelomonocytic leukemia. Patients with tuberous sclerosis can develop hydrocephalus secondary to subependymal giant cell astrocytoma and seizures.
Deterrence and Patient Education
The patients with these syndromes should be educated that there is no cure for these conditions and we can only provide symptomactic management. Genetic counseling should be offered prior to conception.
Pearls and Other Issues
- Neurocutaneous syndromes present with manifestations in the nervous system and the skin
- Neurofibromatosis and tuberous sclerosis complex inherited in an autosomal dominant pattern, with 100% penetrance, but with variable expression
- Neurofibromatosis and tuberous sclerosis are clinical diagnoses.
- The management of neurocutaneous syndromes is surveillance, prevention, and treatment of complications
Enhancing Healthcare Team Outcomes
For a general pediatrician who manages health surveillance of patients, it is imperative to be knowledgeable about the signs and symptoms of neurocutaneous syndromes. Once a clinician suspects a patient might have a neurocutaneous syndrome, it becomes vital for the general practitioner to coordinate the surveillance of these patients' health; this means assuring that the patient will have the appropriate follow up with an interprofessional team, including neurologists, developmental pediatricians, surgeons, ophthalmologists, audiologists, dermatologist and more. It is also essential to have appropriate cognitive function surveillance and to assure that the patients will receive all the educational services available and needed for them to have the best outcome in their education and development.