Introduction
The lungs are a pair of primary organs of respiration, present in the thoracic cavity beside the mediastinum. They are covered by a thin double-layered serous membrane called the pleura.
The respiratory system consists of two components, the conducting portion, and the respiratory portion. The conducting portion brings the air from outside to the site of the respiration. The respiratory portion helps in the exchange of gases and oxygenation of the blood.
The conducting portion of the respiratory system includes the nose, nasopharynx, larynx, trachea, and a whole series of successive narrowing segments of bronchi and bronchioles. The conducting portion end at the terminal bronchiole. The respiratory portion begins from the respiratory bronchiole and continues with the alveolar ducts, alveolar sacs, and finally ends at the alveoli where the significant exchange of gases takes place. The branching pattern of these conducting passages looks like the branching of a tree and hence called the tracheobronchial tree [1].
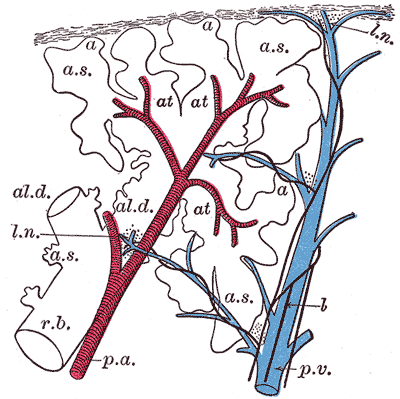
The right lung has three, and the left lung has two lobes. Each is aeriated by a secondary (lobar) bronchus. The lobes are further divided into smaller pyramidal shaped sections called the bronchopulmonary segments. There are ten bronchopulmonary segments in each lung with their apex directed towards the hilum, and each is aeriated by a tertiary (segmental) bronchus [2].
The alveoli are the structural and functional units of the respiratory system. There are around 300 million alveoli in an adult human amounting to approximates 80 square meters of surface area for the gaseous exchange [3].
The lungs are an essential component of the pulmonary circulation where the deoxygenated blood pumped by the right ventricle of the heart is gushed through the pulmonary arteries to alveolar-capillary beds of the lung for gaseous exchange. The oxygenated blood from the capillaries of the lungs is returned to the left atrium by the four pulmonary veins.
Issues of Concern
These are not actually the issues of concern but some of the unique features of the lung.
- The early prenatal development of the lungs depicts the development of an exocrine tubule-alveolar gland. Initially, it begins an offshoot from the endoderm of the foregut as a lung bud. Later, branches to form the tracheobronchial tree and canalizes. This is precisely how the glands develop. Hence initial development of the lung is said to be pseudo-glandular development [4].
- The lung alveoli start developing in the prenatal stage and very important for a live birth with the active secretion of surfactant from its type II pneumocytes. Nevertheless, 95% of alveoli are formed postnatally during the first eight years of life, that too a majority being in the first three years.
- The lymphoid tissue of the tracheobronchial system is modified to contain diffuse, aggregated, and solitary lymphatic nodules specialized to be called bronchus-associated lymphoid tissue (BALT) [5][6]. This is important for the hypersensitive reaction, which occurs with respiration. They also have a significant role in the defensive mechanism from the exposed microorganisms form, reaching the lungs through the inhaled air.
Structure
The conduction portion of the lung begins at the trachea and extends to the terminal bronchioles. Outside the lungs, the conduction system consists of the nasal cavities, nasopharynx, larynx, and trachea. Within the lungs, the conducting portion spits into paired main bronchi. The bronchi begin as a branching pattern, splitting next into lobar (secondary) bronchial branches and then again into segmental (tertiary) bronchi. The tertiary bronchi continue to divide into small bronchioles where the first change in histology takes place as cartilage is no longer present in the bronchioles. The end of the conduction portion of the lungs is at the final segment called the terminal bronchioles. The terminal bronchioles open into the respiratory bronchioles [7]. This is the start of the respiration portion of the lung.
The conducting portion provides the pathway for the movement and conditioning of the air entering the lung. Specialized cells collaborate to warm, moisturize, and remove particles that enter. These cells are the respiratory epithelium and comprise the entire respiratory tree. Most of the respiratory epithelium is ciliated pseudostratified columnar epithelium. The following five types of cells are in this region:
- Ciliated cells
- Goblet cells
- Basal cells
- Brush cells
- Neuroendocrine cells
The ciliated cells are the most abundant. They control the actions of the mucociliary escalator [8], a primary defense mechanism of the lungs that removes debris. While the mucus provided by the goblet cells traps inhaled particles, the cilia beat to move the material towards the pharynx to swallow or cough out.
Goblet cells, so named for their goblet-shaped appearance, are filled with mucin granules at their apical surface with the nucleus remaining towards the basilar layer. Goblet cells decrease in number as the respiratory tree gets progressively smaller and are eventually replaced by club cells (previously Clara cells) when they reach the respiratory bronchioles.
The basal cells connect to the basement membrane and provide the attachment layer of the ciliated cells and goblet cells. They may be thought of as the stem cells of the respiratory epithelium as they maintain the ability to potentiate ciliated cells and goblet cells [9].
Brush cells, occasionally referred to as type III pneumocyte cells are sparsely distributed in all areas of respiratory mucosa. Brush cells may be columnar, or flask-like and are identified by their short microvilli covered apical layer–resembling a push broom or appropriately, a brush. No function has been officially assigned to the brush cells though there are many proposed mechanisms. One popular proposal suggests they have a chemoreceptor function, monitoring air quality, due to their association with unmyelinated nerve endings. [10]
The bronchial mucosa also contains a small cluster of neuroendocrine cells, also known as Kulchitsky cells [11]. They have neurosecretory type granules and can secrete several factors. This includes catecholamine and polypeptide hormones, such as serotonin, calcitonin, and gastrin-releasing factors (bombesin). Like brush cells, these neuroendocrine cells make up only a small portion of mucosal epithelium, around 3%.
Within the bronchial submucosa are submucosal glands. These glands are composed of a mixture of serous and mucinous cells, similar to salivary gland tissue. The secretions are emptied into ducts and then on the bronchial mucosa. Older individuals may show oncocytic metaplasia of these glands. Smooth muscle bundles are present at all levels of the airway to allow for regulation of airflow. There are progressively fewer smooth muscle fibers progressing from bronchi to alveoli.
Function
Respiratory functions of lungs:
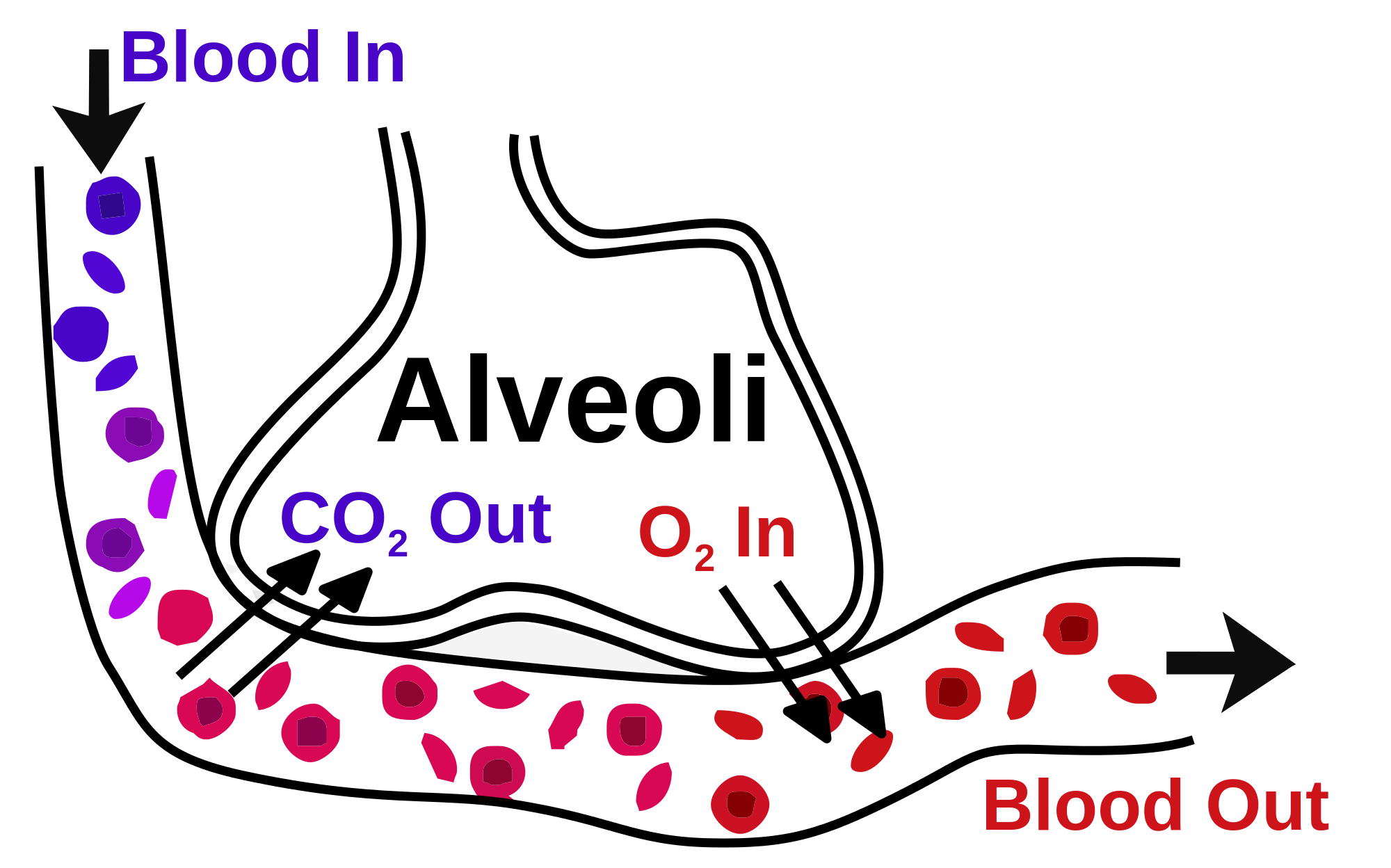
- Respiration: The lungs are the primary organs of respiration, where the exchange of gases takes place. In the alveoli, which are the functional units of lungs, the oxygen is taken up, and carbon dioxide is removed from the bloodstream through the alveolar-capillary bed [12].
- Air conditioning: The function of conducting part is not only to lead the air but also acts as an efficient air conditioner. This conditioning is done by warming/cooling the inhaled air to bring it to the level of body temperature, humidifying the air, and also the removal of all the foreign particles present in it. The removal of foreign particles like dust, bacteria, virus, etc., is done by mucous secretion, which traps the suspended particles and beating of the cilia, which clears the mucus from the respiratory passage [7].
Non-respiratory functions of lungs:
Even though the lungs are primarily for respiration, studies indicate that they have many non-respiratory functions. Some of the few important ones are mentioned below.
- Converting an inactive chemical precursor into its active form, like turning angiotensin-I into angiotensin-II, which help in raising of blood pressure [13].
- It is also an essential site for degrading/inactivating important vasoactive chemical mediators like bradykinin, serotonin, and norepinephrine
- The bronchial mucosa also contains a small cluster of neuroendocrine cells [14], also known as Kulchitsky cells that can secrete several factors, including catecholamine and polypeptide hormones, such as calcitonin, serotonin, and gastrin-releasing factors (bombesin)
- Pulmonary epithelium acts as the first line of defense for the inspired air [15].
Tissue Preparation
Proper ethical approval is obtained before the collection of the lung tissue. The lung is identified, dissected en-block, weighed, and labeled. Later the lung is perfused with 10% formalin through the trachea to the physiological peak inspiration level. Underinflation or over inflation should be prevented. This helps in proper assessment without any artifacts and over/under the judgment of the tissue structure. All the lobes of the lungs are identified searched for any lesions. The tissue is washed well, fixed in formalin for almost 24 hours. Once the tissue processing is completed, it is taken to the next level of tissue embedding. The lung organ should be placed in the tissue cassette with the ventral side facing the tissue cassette. The dorsal side of the lung will be facing the open/upper side. This position guarantees the appropriate tissue section level. Post fixation tissue trimming is a crucial component to get the best sections. If the identified lesions are too big or small, they can be isolated and embedded separately. It is essential to include the sections of related lymph nodes for histological evaluation, which will help in turn to understand the extent of metastasis of the lung tumors [16].
Histochemistry and Cytochemistry
Histochemistry of the Lung
Histochemical analyses of the lung can be carried out using lectin histochemistry (LC) or immunohistochemistry (IHC). Both methods can be performed on tissue slides. The normal lung cells can be typed as type I and type II pneumocytes based on histochemical analyses using lectin typing (glycotyping) [17]. The alveolar macrophages in the normal lung are positive for many N-linked saccharides, namely, N-acetylgalactosamine, N-acetylglucosamine, terminal β-D-galactose, and sialyl groups. Some specific lectins can be used as a marker for cell types, such as Dolichos biflorus agglutinin for bronchial epithelial cells, Triticum Vulgaris (succinylated) for type I pneumocytes and Hippeastrum hybrid or Maclura pomifera lectins for type II pneumocytes. Alveolar macrophages are anti-CD68 positive, and alveolar lining show positivity for cytokeratin.
Further, IHC and LC can provide substantial insights into obstructive and restrictive pathologies as well as in lung cancer diagnosis. Adenocarcinomas can be identified by differentiation markers that include TTF-1 and NapsinA. Both these markers are expressed in more than 85% of the cases [18]. Metastasis can be confirmed by IHC staining to identify primary tumor tissue of origin. Distinct expression of CK7 and CK20 profiles, in addition to the absence of markers commonly expressed in primary lung cancer, can signify metastatic cancer. With the advent of cancer treatment through tyrosine kinase inhibitor (TKI), actionable gene mutations can be screened in lung cancer patients [19]. EGFR mutation and ALK translocation are the most effectively targeted oncogenes in non-small cell carcinomas and are now considered standard treatment procedures presently [19].
Cytochemistry of the Lung
Cell analyses of the lung tissue can be carried out using electron microscopy and have recently included flow cytometric assays. Assessment of non-specific esterases: alpha-naphthyl acetate esterase (ANAE) and butyrate esterase (BUT), chloroacetate esterase (CHL), acid phosphatase (ACP), intracellular glycogen (PAS reaction), lipids (Sudan black B reaction-SBB) and iron (Perl's reaction) can be performed by a semiquantitative cytochemical method to identify diseases of the lung [20]. ECM remodeling is the basis of pathologic changes in obstructive pulmonary diseases, and collagen alteration provides insight into the disease. The second-harmonic generation (SHG) technique used to quantify collagen has demonstrated effect analysis of lung diseases, with a biochemically distinct presentation of organization of collagen in asthma, COPD and idiopathic pulmonary fibrosis (IPF) [21]. Alveolar macrophage modulations using chloroacetate esterase (CHL) and Perls' reaction (for estimation of intracellular iron) can confirm the presence of non-small cell lung cancer. The typical increase in CHL and iron in alveolar macrophages indicates non-small cell lung cancer [10]. Similarly, Galectin-3 (Gal-3) can be used as a biomarker in human pulmonary fibrosis as the levels are observed to be elevated in this disease [22].
Microscopy, Light
As mentioned before, the conducting portion is up to the terminal bronchiole. Beyond that with the beginning of respiratory bronchiole is the respiratory part of the respiratory system. The respiratory portion of the lung consists of respiratory bronchiole, alveolar duct, alveolar sac, and finally alveoli where actual respiration takes place.
In the conducting zone, the air is moistened, warmed, and filtered before it reaches the start of the respiratory region at the respiratory bronchioles. The respiratory zone is where gas exchange occurs, and blood is oxygenated in exchange for carbon dioxide. As the respiratory tree transitions from the conducting zone at the terminal bronchioles, goblet cells diminish as club cells increase, and the cartilage present in the conducting region is absent once it reaches the respiratory bronchioles.
The acinus is directly distal to the terminal bronchioles and which signals the beginning of the respiratory part. The acinus is composed of respiratory bronchioles, alveolar ducts, and alveolar sacs. It is roughly spherical, resembling a bunch of grapes. Each respiratory bronchiole gives rise to several alveolar ducts and alveolar sacs, giving it the characteristic grape bunch appearance. The alveolar sacs are the ends of the respiratory tree and the site of gaseous exchange.
Alveolar epithelium is composed of type I pneumocytes, type II pneumocytes, and the occasional brush cells. Also present in the alveolar walls are the club cells and alveolar macrophages. The alveolar walls contain the pores of Kohn [23][24], which allow communication between adjacent alveoli. This allows air to flow from one alveolus to another, which may be beneficial if there is any blockage preventing air from entering alveoli through a direct route.
Type I pneumocytes make up roughly 90% to 95% of the alveolar epithelium. They are flat, squamous epithelia that resemble plate-like structures that allow gas exchange. Their fragile membrane allows for easier gas permeability between the alveoli and the blood vessels. Despite being the primary structures of respiration, they cannot replicate and are very susceptible to toxic injury.
Type II pneumocytes make up much of the remaining cell type in the alveoli, accounting for nearly 5-10%. Despite their low number, they are vital as they secrete pulmonary surfactant. The surfactant is necessary to maintain an open airway. It lowers the surface tension and prevents the alveoli from collapsing upon themselves during exhalation. By histology, these cells have foamy cytoplasm, which results from the surfactant that is stored as lamellar bodies. Type II pneumocytes are also mitotically active and can replace the easily damaged type I pneumocytes. Type II pneumocyte cells can be recognized by their rounded shapes that bulge into the alveolar space.
Alveolar macrophages (or dust cells) may be free within the alveolar space or sometimes connected to the alveolar wall. If particles make it down to the acinus, the macrophages are the last defense and janitors of the respiratory epithelium. The black staining seen in the lungs of smokers results from macrophages cleaning and sequestering particles that make their way inside.
The lungs are covered by the serous membrane, the pleural membrane, which has two layers - the parietal and the visceral layer. The visceral pleura of the lung is lined by a mesothelial layer with underlying connective tissue and elastic fibers. An elastin stain may be used to identify the elastic layer.
Microscopy, Electron
In this section, let us get into some more detailed study of a few of the essential structures of the lung, which can be appreciated better under an electron microscope.
Respiratory Bronchiole:
This is the region of transition between conducting and respiratory portions (where the exchange of gases begins). Structurally respiratory bronchiole is very similar to terminal bronchiole, except its walls are interrupted by numerous sac-like alveoli for gaseous exchange. It is lined by ciliated cuboidal epithelium. The cilia may be absent in more distal portions. A small number of non-ciliated Clara cells are also present. These Clara cells become dominant cells in the distal part. The epithelium is devoid of goblet cells. Underlying tissue consists of smooth muscle and elastic fibers. Distally the respiratory bronchioles divide, become narrower, and the number of alveoli increases.
Clara cells have three functions,
- Produce one component of Surfactant,
- Act as stem cells and
- Contain enzyme systems that detoxify noxious substances
Alveolar ducts:
Respiratory bronchioles divide distally to form alveolar ducts. Alveolar ducts do not have walls of their own but are created by several openings of alveoli. These terminate into clusters of alveoli known as the alveolar sac, which opens into the atrium and finally into alveoli. Alveolar ducts are surrounded by small aggregations of smooth muscle cells, collagen, and elastic fibers. Sphincter-like smooth muscles (knob) embedded in type III collagen around respiratory bronchioles and alveolar ducts regulate air movements within alveoli. Smooth muscle totally disappears at the distal end of alveolar ducts. Rich matrix of elastic and reticular fibers provides the only support for duct and alveoli
Alveoli:
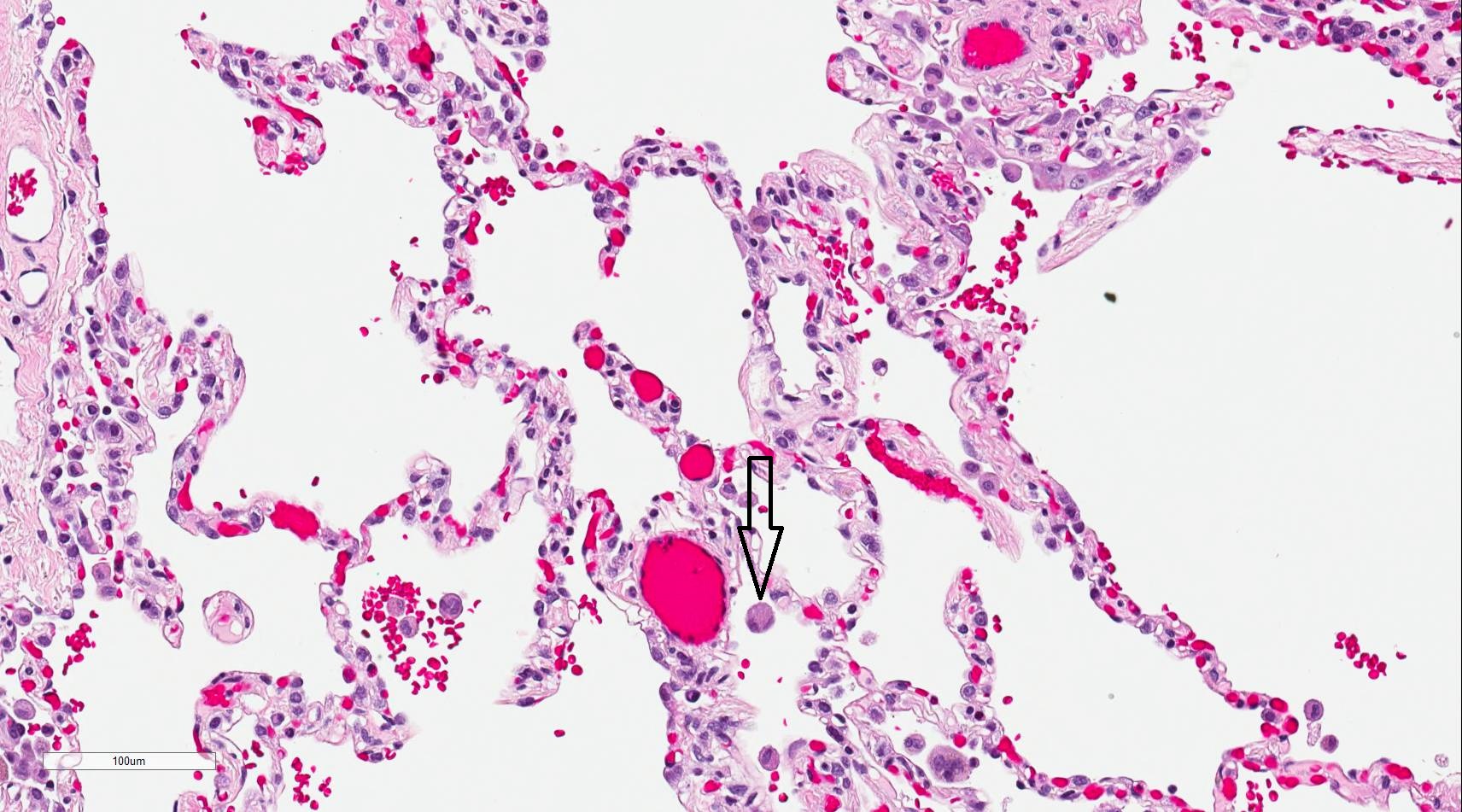
The alveoli are the specialized air-sacs (outpouching) of 200 µm diameter. These are the structural and functional unit of the respiratory system. These are the primary site of exchange of oxygen and carbon dioxide. There are 300 million alveoli in the lungs, and these give approximately 140m surface area for the exchange of gases. The alveoli are responsible for the spongy nature of the lung. These alveoli are lined by flattened epithelial cells called pneumocytes with a single opening. The alveolar wall or septum is made up of three tissue components: surface epithelium, supporting tissue, and an extensive network of continuous capillaries. Centrally it has capillaries surrounded by a vibrant network of elastin, reticular, and collagen fibers with a layer of squamous epithelial of two adjacent alveoli on either side. In certain places, the basement membrane of the capillary endothelium comes in direct contact with the basement membrane of the surface epithelium of alveoli, with the absence of supporting tissue, thus reducing thickness (0.1-1.5µm) for better exchange of gases. Hence air in alveoli is separated from the blood in the capillary by three components, surface lining and cytoplasm of alveolar cells, fused basal laminae of closely apposed alveolar, and endothelial cells and their cytoplasm. This is called the blood-gas barrier (air blood barrier). Capillaries are continuous with its endothelial cells, which are extremely thin due to the clustering of nuclei and other organelles, increasing the efficiency of exchange. Occasionally small openings, alveolar pores (of Kohn) (10-15µm in diameter) are seen which equalize air pressure within alveoli and allow air movement between alveoli in case of the bronchiole obstruction. Elastic fibers enable alveoli to expand during inspiration and contract passively during expiration. The reticular fibers serve as supportive structures that prevent over-distention and damage to delicate capillaries and alveolar septa. Two types of epithelia form a continuous lining around each alveolus. They are type I pneumocytes (alveolar lining cells) and type II pneumocytes.
Type I Pneumocytes (Alveolar lining cells)
The type I Pneumocytes are simple squamous cells that are highly attenuated with a dense, small, and flattened nucleus. These cover most of the surface area, approximating around 95-97% of the total surface area. Golgi complex, endoplasmic reticulum, and mitochondria are grouped around the nucleus, leaving a large area of cytoplasm free of organelles, thus reducing it to a fragile blood-air diffusion barrier (25nm). The thin cytoplasm shows numerous pinocytotic vesicles. The surfactant lines the luminal surface. A basal lamina covers the adluminal side of these cells. The adjacent cells are connected by tight (occluding) junctions, which prevent leakage of tissue fluid into the alveolar lumen. During fetal development, the surfactant appears in the last few weeks of gestation and coincides with the appearance of lamellar bodies in the type II cells.
Type II Pneumocytes (Great alveolar or septal cells)
The type II Pneumocytes are the cuboidal cells grouped in 2-3, large, a central, and plump nucleus with dispersed chromatin and prominent nucleoli. They occupy about 3-5% of the surface area of alveoli interspersed among type I cells with which they have occluding and desmosomal junctions. The apical surface is dome-shaped and shows numerous small microvilli associated with surfactant secretion. These type II pneumocytes secrete Surfactant, a surface-active material that reduces surface tension, thus preventing alveolar collapse during expiration. The mitotic activity of the lining cells is 1% per day and can differentiate to type II, as well as type I pneumocytes in response to damage to alveolar lining epithelium. The cytoplasm has abundant rough endoplasmic reticulum, well developed Golgi apparatus, free ribosomes, and a moderate amount of elongated mitochondria. A typical feature of these cells is the presence of lamellar bodies. These lamellar bodies are vesicles containing phospholipid (dipalmitoyl phosphatidylcholine) (1-2µm). These, when discharged by exocytosis into alveoli, spreads on alveolar surface and combines with other carbohydrate and protein-containing secretory products (some secreted by Clara cells) to form surfactant, which is a tubular lattice of lipoprotein known as tubular myelin, which overcomes effects of surface tension. The reduction of surface tension means a reduction of work of breathing. The surfactant is not static but continually being turned over and removed. The removal is by pinocytotic vesicles of type II pneumocytes, macrophages, and type I pneumocytes.
Alveolar Macrophage (Dust cells):
The alveolar macrophages are derived from blood monocytes and sometimes by mitotic division of macrophages of the lung. They contain numerous secondary lysosomes and lipid droplets. They phagocyte and remove unwanted materials such as inhaled particulate matter (carbon), dust, and bacteria. They are present free within alveolar spaces and some in inter-alveolar septa (spaces). One hundred million macrophages daily migrate to bronchi. The phagocytosed macrophages get trapped in mucus, transported by ciliary action to the pharynx, and come out in sputum. Some alveolar macrophages also go via lymphatics to hilar lymph nodes. Industrial lung disease like silicosis is because of inhalation of silica into air sacs (as tiny particles) that are phagocytosed by macrophages. Silicated macrophages stay for a long time and convert silica into silicic acid, which stimulates the proliferation of fibroblasts and collagen, leading to fibrosis of lung and node. A particular form of silica, like asbestos, when inhaled extensively stimulates lung fibrosis producing asbestosis and sometimes malignancy of pleura (mesothelioma). Sometimes macrophages phagocytose extravasated RBCs in alveoli (especially in conditions like pulmonary congestion and congestive heart failure). These erythrocyte phagocytosed macrophages are called heart failure cells.
Pathophysiology
The pathologies associated with the lung are diverse, with significant variations in disease presentation. This is primarily due to the exposure of this organ to the outside environment. Lung pathologies can be broadly classified as obstructive lung diseases or restrictive lung diseases.
This includes bronchial diseases, infectious diseases, interstitial lung diseases, neoplasm, vascular diseases, congenital abnormalities, etc. The destruction of lung parenchymal tissue presents chronic obstructive pulmonary disease and emphysema due to chronic inflammation [25]. Morphologically they lead to an increase in size and number of small fenestrae in alveolar walls, fibrovascular trabeculae breakdown, and remodeling of acini leading to airspace enlargement [26].
In some obstructive diseases such as bronchitis, hyperplasia of goblet cells occurs, while in bronchiectasis, the bronchi are markedly dilated. Restrictive disorders, on the other hand, are marked by the fibrous deposits that restrict lung function. Interstitial restrictive lung diseases are characterized by inflammation or scarring of the lung tissue or filling of the air spaces with exudate and debris. Extrapulmonary restrictive diseases show thickening of alveolar septa and epithelium and are associated with an endothelial injury.
In its acute phase, restrictive lung disease demonstrates endothelial damage. In some disorders, they also show epithelial damage with fibrosis of the exudate and expansion of the interstitium through generalized fibrosis. Chronic restrictive lung diseases are marked by diffuse interstitial changes that are more prominent than the morphological changes. Many of the chronic restrictive lung diseases, particularly in later stages, are characterized by interstitial fibrosis leading to a classic honeycomb-like appearance of the lung.
Clinical Significance
Infant Respiratory Distress
Infant respiratory distress is the leading cause of death in premature babies. Type II pneumocytes produce surfactant starting around 20 weeks gestation, but it is not fully secreted until nearly 30 weeks of pregnancy. Without ample surfactant, premature infants cannot overcome the collapsing surface tension in the respiratory alveoli. Physicians hope to prevent infant respiratory distress when a patient goes into premature labor by offering the parent glucocorticoids.[27] Glucocorticoids stimulate the production of surfactant in the fetus and may increase its production enough to help the infant overcome any potential respiratory distress. Testing for fetal lung maturity may be performed on the amniotic fluid by several methods, including the measurement of phospholipids in amniotic fluid (phosphatidylglycerol (PG) or lecithin-sphingomyelin ratio) and the lamellar body counts (LBC).
Emphysema
The most common cause of emphysema is smoking; although, it can be caused by repetitive inhalation of any foreign particulate material. Emphysema, or chronic obstructive pulmonary disease (COPD), is characterized by poor airflow and difficulty exhaling because of narrowing bronchioles and the destruction of the alveolar wall. The collapse of the alveoli results in a significant loss of surface area for gas exchange.
Healthcare professionals assume that the destruction of the alveolar wall is a result of excessive lysis of elastin in the interalveolar septum. The abundance of macrophages and neutrophils that migrate to the acinus due to an increase in particulate bring an equal rise of elastase and other proteases. Alpha 1-antitrypsin deficiency also causes emphysema because of an increase in elastin; however, in this disease, it is because the deficient antitrypsin usually inhibits elastin.
Cystic Fibrosis
Cystic fibrosis may also cause chronic obstructive pulmonary disease. It is an autosomal recessive disease caused by a mutation to the CFTR gene on chromosome 7. This gene controls the Cl- channel protein involved in a variety of cells, including goblet cells in the lungs. The defective Cl-channel affects the viscosity of the mucus in the lungs. Thickening is due to increased absorption of sodium (Na) and water from the lumen. The thickened mucus disrupts the mucociliary escalator filtration function of the lungs resulting in obstruction. One of the supportive treatments for cystic fibrosis breaks the disulfide bonds found in mucous plugs, thinning out the sputum so it can be pushed out by the respiratory cilia.[28]
Heart Failure Cells
In heart failure, the heart's inability to move blood efficiently results in congestion of the lungs. The increase in pressure of the blood in the pulmonary vasculature results in erythrocytes passing into the alveolar septum. The erythrocytes are promptly taken up by resident alveolar macrophages. As the macrophages engulf any red blood cells present, they are filled with hemosiderin and take on a brown granule appearance viewable under light microscopy with staining.[29] Hemosiderin-laden macrophages are more accurately called siderophages and are not specific to a particular disease but may be present whenever blood cells enter the alveolus.
Tumor Staging
When staging primary carcinoma of the lung, it is crucial to identify the invasion of the elastic layer of the visceral pleura. This finding will increase the T stage of the tumor. Since the elastic layer is difficult to visualize with routine stains, a special elastin stain may be used to demonstrate this finding.